Doctorate Course
25 Oct -5 Nov 2010
Slides:
1. Introduction
2. Databases_1
3. Databases_2
4. Alignments
5. DotPlots
6. MSA
7. Pymol_slides
ANALISYS OF BIOLOGICAL DATABASES
Outline
Bioinformatics and Internet. Genomes and sequence analysis. Proteomes and sequence analysis. Structural Bioinformatics
Installing software
The specific software used along these practices is linked in the corresponding exercises. Some of them are portable application, but others require typical installation. Start with the installation of Python 2.6 in your PC and locate the IDLE program to edit (and run) python scripts; use this script example. Just open and have a look. Take care of the computer you are using because the main drive (C:\) is frozen, so that all files or installed programs are lost upon rebooting, and you will need to reinstall the program.
Exercises
1.- Tools for bioinformatic
Practice 1.1: HTML language.
Create a web page locally in html code with all names and links of the databases related with Molecular Biology, Structural, Cell Biology, etc. you will visit along these practices. Sort all these database in alphabetical order and add a brief description of the database. At the end of the practices, this file will have near 50 entries. As an example:
ADAN:
Prediction of Protein-Protein Interaction of Modular Domains
SBMC:
Structural Biocomputing and Modeling Center at IBMC-UMH
SMART: simple modular
architecture research tool, etc., etc.
Tricks: The simplest way to edit an html page is to create a text document in MS-Word (*.doc) or in OpenOffice (*.odt) text document and saving in *.html format. Visualize the page in the browser to see the final presentation.
Practice 1.2: Transfer files to a remote computer.
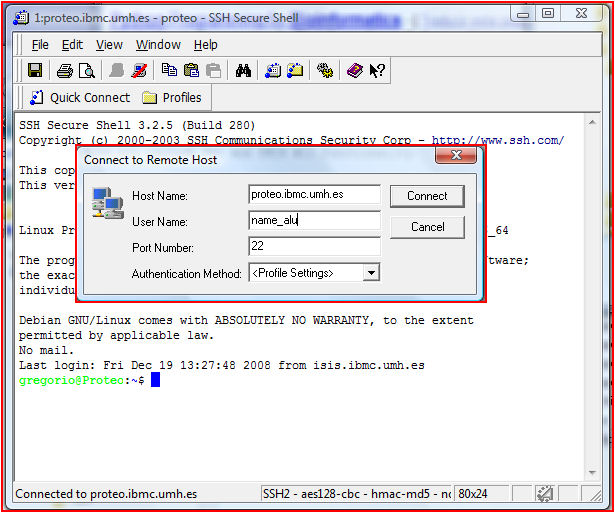
Connect to a remote computer to the Structural Biocomputing and Modeling Center (SBMC) at IBMC-UMH and transfer a file. For this and many other practices you need a login and a password. Connect to server "proteo.ibmc.umh.es" using your own login and password provided by your supervisor.
Tricks: Use the
SSH Client
program: File/Quick Connect...
and write the server name and your login (figure left, insert). Then you will be asked for a
password. You will see a shell with the confirmation that you
have entered in the server (figure left). The command line can be used to run command
or programs. Try the following commands: whoami, ls, ls -al, pwd, top (ctrl+c
to break).
Now you can transfer a file to an appropriate folder
in two steps:
1. At the prompt type "mkdir" to create the folder: $mkdir
folder_name. Now change to the new folder: $cd folder_name. Check your new location: $pwd.
Tipically you will want to have a folder structure like this:
..../RESULTS/P1/P1.2
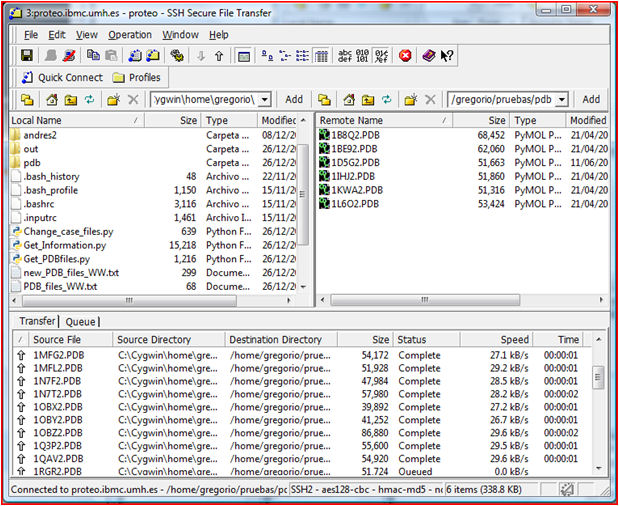
2. Go to your SSH shell and select Windows/New File
Transfer. A new window appears (figure right). Left panel is your local
machine while right panel is the remote one. Select the appropriate
folder either in the local or remote computers. As an example download
locally the compressed file
GetPDBfiles
and transfer it to the remote server. For this just select (left+click)
the file in the left panel and drag to the desired folder in the right
panel.
Practice 1.3: Run programs in a remote computer. Run prochek to check a structure file. Later use the program ps2pdf to transform *.ps to *.pdf.
Connect with ssh to proteo.ibmc.umh.es server (login: name.familyname pass: nick) and run the program prochek to check the structure Model1.pdb at 2.1 Å resolution. Visualize the Ramachandran plot. Could we run prochek from a web server?
Tricks: Connect to the machine and make an appropriate folder (remember: no spaces, no weird characters). Transfer the file to this folder. Run prochek to see the command line format and run the program. Use the following format: %prochek Model1.pdb 2.1. The results are "*.ps". Transform to pdf using the program "ps2pdf"; focus on Model1_01.ps and download the result from the server to your computer. Then store the result correctly in the server.
Practice 1.4: Capture results from the screen.
A useful way to get and store the results obtained is the screen capture. You will use this utility along these practices. Capture any part of your actual screen and paste it in a word processor. Alternatively you can print the page in pdf format.
Tricks: Use the portable software Lightscreen for captures. For printing in pdf use DoPdf.
2.- Scripting & Database Management
Practice 2.1: Get or update structural information from a database: SMART. In this practice we want to know which and how many structures are available for certains domains in SMART. These domains are WW, SH3, SH2, PDZ. Imagine you want to get a file with all the names of the structure files, but you already have some of these names.
Tricks: This job is quite tedious. It is important to realize that the scriptting programs are invaluable help to automatize work. Use the python script SMART_domain_checker. Edit the file locally in windows with python IDLE, NotePad or EditPlus and follow the instructions inside. Transfer the file and run the script in the SBMC server "proteo.ibmc.umh.es". Run the script four times, one for each domain. Check the text content of the *_XX.txt files 'before' and 'after' the use of the script, as well as the 'new_PDB_files_XX.txt' files that are generated by the script.
Practice 2.2: Get all pdb structure files from PROTEIN DATA BANK. Now we want to get all pdb files found in Pratice 2.1.
Tricks: Again the job is tedious. Use the python script Get_PDBfiles. Copy the script to "proteo" in an appropriate folder (no edition is needed) containing the list of desired *.pdb files obtained in exercise 2.1. Execute script as follows:
$python GetPDBfile.py filename_list.txt (enter)
Practice 2.3: Change all pdb file names to uppercase. This is just for convenience. Most of the times several terminology problems are avoided if names of structures, sequences, etc. are homogeneous.
Tricks: Use python script Change_case_files. Edit the file locally in windows with python IDLE, NotePad or EditPlus and follow the instructions inside. Transfer the file and run the script in the SBMC server "proteo.ibmc.umh.es".
Practice 2.4: Get Information for pdb files. This practice is conceptually very simple: Once you have a list of pdb files from a given domain, you want to extract as many information as possible from each domain, namely: 'Swiss-prot-ENTRY_NAME', 'Swiss-prot-AC_number', 'Sequence domain', 'Sequence ligand', 'Putative partners', 'Cellular localization', 'Drugs', 'Phosphorylated residue type', 'Experimental Technique', 'Protein Source', 'Title', 'Resolution', 'MedLine_ID', 'Reference', 'Protein family', 'Keywords', etc. Visit:
http://www.uniprot.org/uniprot/Q04439 (Myo5 protein from S. cerevisiae) to check that all these information is easily accesible from UNIPROT. However, the automatation of this data extraction is a bit more complicated.
Tricks: Download python 2.6+modules modules and install them. Download the file Get_Information, modify the necessary lines and run the python script Get_Information.py in Windows (not Linux) using the IDLE editor. Some example files (cleaned and original pdb files) are provided. The domains belong to the PDZ family.
3.- Managing Protein Databases
Practice 3.1: Try to find an alive database (with its corresponding home server address and the date of the latest update) dealing with: [solution]
- nucleic acid sequences (in Japan)
- microarray data
- mass spectrometry data repository
- protein protein interaction data
- rat enamel 2D gel electrophoresis
- CFTR mutations
Tricks: You can search databases with Google or with the ExPASy Life Science Directory
Practice 3.2: Human erythropoietin (EPO) in different protein sequence databases. Find, if it exists, one entry corresponding to human erythropoietin protein sequence in the following protein sequence databases: [solution]
- NCBInr http://www.ncbi.nlm.nih.gov/
- RefSeq http://www.ncbi.nlm.nih.gov/RefSeq/
- UniProtKB http://www.uniprot.org/find sequence(s) in UniProtKB/Swiss-Prot and sequence(s) in UniProtKB/TrEMBL
- PIR-PSD http://pir.georgetown.edu/pirwww/dbinfo/pir_psd.shtml
- UniParc http://www.uniprot.org/
- UniRef50 http://www.uniprot.org/
- Ensembl http://www.ensembl.org/Homo_sapiens/searchview
- IPI IPI query
Find the UniProtKB/Swiss-Prot entry corresponding to:
- RefSeq NP_036231 - GI:584682
Tricks: You can use the
query tool provided by each database.
You can use
SRS
You can use the crosslinks (if they exist) to go from one database to
another...
You can use the
ID/AC mapping tool provided by UniProt
Practice 3.3: Searching all the protein sequences for the gene "ken and barbie" with text search tools
- Enter "ken and barbie" in the text search box of UniProt website
- How many sequence do you get? Are there mainly reviewed or unreviewed?
- Do the same search at NCBI
Tricks: [solution]
Practice 3.4: Different looks and tools for the same entry....
- Starting with the UniProt server: (http://www.uniprot.org/):
- Look for the amino acid sequence of human carbonic anhydrase 2.
- Get the corresponding nucleic acid entries in EMBL and GenBank: try to find a nucleic acid sequence derived from genomic DNA sequencing and another one derived from cDNA sequencing.
- From the UniProtKB/Swiss-Prot entry, look at the data available for variant Pro-92 and in particular its position in the 3D structure (Use the “Jmol viewer”).
- Starting with the NCBI server: (http://www.ncbi.nlm.nih.gov/):
- Look for the amino acid sequence of human carbonic anhydrase 2 using ENTREZ Protein.
- Find the UniProtKB/Swiss-Prot entry and as above:
- Get the corresponding nucleic acid entries in EMBL and GenBank.
- Find the data available for the variant Pro-92.
Tricks: [solution]
Practice 3.5: Browsing a database. Search in UNIPROT (http://www.uniprot.org) the sequence of the botulinum toxins (types A to G) from Clostridium botulinum. Optimize the search so that the query produce 10 results or less. Extract and download the sequence in fasta format. [solution]
Tricks: Take advantage of the facilities in the UNIPROT server. Download results in fasta format. This format is as follows:
>bontoxilysin A
MPFVNKQFNYKDPVNGVDIAYIKIPNAGQMQPVKAFKIHNKIWVIPERDTFTNPEEGDLNPPPEAKQVPVSYYDSTYLSTDNEKDNYLKGVTKLFERIYSTDLGRMLLTSIVRGIPFW..
>bontoxilysin B
MPVTINNFNYNDPIDNNNIIMMEPPFARGTGRYYKAFKITDRIWIIPERYTFGYKPEDFN
KSSGIFNRDVCEYYDPDYLNTNDKKNIFLQTMIKLFNRIKSKPLGEKLLEMIINGIPY...
Practice 3.6: Search all proteins containing SH3 domains in S. cerevisiae and S. pombe. Use SMART as the source database and download protein architecture, whole protein sequence in fasta, and SH3 protein sequence in fasta, as well.
Tricks: Visit SMART and select Normal Mode. Take advantage of the query capabilities and the taxonomic selection.
Practice 3.7: 3D structure database
- Software: Java Virtual Machine.
- Find the 3D structure of 1IWO at PDB.
- Look at the complete coordinates of the entry (by clicking to Download/Display file) and save it in a text file.
- Edit the former text file and try to find a line starting with "DBREF". Look at the cross-reference to UniProtKB/Swiss-Prot.
- Visualize the structure-sequence relationships with Quick PDB.
- Can you identify the transmembrane helices? How are they annotated in the corresponding UniProtKB/Swiss-Prot entry?
- How many PDB entries are there for the lysozyme T4?
Tricks: [solution]
4.- Managing Genomic Databases
Practice 4.1: Browsing genes in genome databases
- Look for the UniProtKB/Swiss-Prot entry of the E.coli gene gutQ.
- Follow the link to EcoGene (EcoGene Database of Escherichia coli sequence and function) and find the chromosomal location.
- Get the next E.coli gene on the same strand.
- Follow the link to UniProtKB/Swiss-Prot.
- Find the subcellular localisation of the protein.
Tricks: [solution]
Practice 4.2: Locating genes in chromosomes
- Find the IL-2R alpha gene in the OMIM database? What is its chromosomal location ?
- View the cytogenetic maps of the regions surrounding the gene loci
- Are there known diseases associated with this gene? What are the associated disorders ?
- From OMIM (IL-2R alpha), follow the cross-reference to Entrez Gene. Have a look to the Reference Sequences (RefSeq).
- Find the corresponding UniProtKB/Swiss-Prot entry
Tricks: [solution]
Practice 4.3: Gene ontology database: structure and validation of the data...
- Look at the definition of ontology in wikipedia
- Go to the Gene ontology consortium (AmiGO)
- Compare the GO terms associated with mouse and human erythropoeitin
- Go to the UniProtKB/Swiss-Prot entry for human erythropoietin: look at the associated GO terms How many terms have been 'manually' attributed to the gene (have a look to the GO evidence tag )?
Tricks: [solution]
Practice 4.4: Comparing two putative splice variants of a human gene. The following two protein sequences, from SwissProt and TrEMBL, map to the same locus on human chromosome 1. They are both derived from RNA sequences that were originally isolated from cancerous tissues.
>EKI2_HUMAN MAVPPSAPQQRASFHLRRHTPCPQCSWGMEEKAAASASCREPPGPPRAAAVAYFGISVD PDDILPGALRLIQELRPHWKPEQVRTKRFTDGITNKLVACYVEEDMQDCVLVRVYGERT ELLVDRENEVRNFQLLRAHSCAPKLYCTFQNGLCYEYMQGVALEPEHIREPRLFRLIAL EMAKIHTIHANGSLPKPILWHKMHNYFTLVKNEINPSLSADVPKVEVLERELAWLKEHL SQLESPVVFCHNDLLCKNIIYDSIKGHVRFIDYEYAGYNYQAFDIGNHFNEFAGVNEVD YCLYPARETQLQWLHYYLQAQKGMAVTPREVQRLYVQVNKFALGPSCVSSTMTASLQCC RVGNRHGEIARLTLSGLFPGVSLLLGSLGPHPEPVLHHRL
>Q96G05 MAVPPSAPQPRASFHLRRHTPCPQCSWGMEEKAAASASCREPPGPPRAAAVAYFGISVD PDDILPGALRLIQELRPHWKPEQVRTKRFTDGITNKLVACYVEEDMQDCVLVRVYGERT ELLVDRENEVRNFQLLRAHSCAPKLYCTFQNGLCYEYMQGVALEPEHIREPRLFRLIAL EMAKIHTIHANGSLPKPILWHKMHNYFTLVKNEINPSLSADVPKVEVLERELAWLKEHL SQLESPVVFCHNDLLCKNIIYDSIKGHVRFIDYEYAGYNYQAFDIGNHFNEFAGVNEVD YCLYPARETQLQWLHYYLQAQKGMAVTPREVQRLYVQVNKFALASHFFWALWALIQNQY STIDFDFLRYAVIRFNQYFKVKPQASALEMPK
Verify that the two proteins can indeed be (nearly) perfectly mapped to the same human genomic region
>HUMAN_TRIMMED_1 GACTTCCCCTGGCCCCTCCTCTACCACTCCCACTCCCTCGCCGGACCCCCCCGCCGGGG CTAGCGTCTGCCGCGGCTCCGAGGGGGTGGGGCTGCTGGGAATGGCTGTGCCCCCTTCG GCCCCTCAGCCGCGCGCGTCCTTTCACCTGAGGAGGCACACGCCTTGCCCGCAGTGCTC ATGGGGCATGGAGGAGAAGGCGGCGGCCAGCGCCAGCTGCCGGGAGCCGCCGGGCCCCC CGAGGGCCGCCGCCGTCGCGTACTTCGGCATTTCCGTGGACCCGGACGACATCCTTCCC GGGGCCCTGCGCCTCATCCAGGAGCTGCGGCCGCATTGGAAACCCGAGCAAGTTCGGAC CAAGGTAGCGGAGTGGGCGCGGGGCCGAGGATGGGGTCCTGCCAGGGCTGCCAGGGGGA TGCGGGGACCCAGTCCTCGCATCCTTCCGGGTGTCAGTCCCAGGCAAATGAGCACCTCC TGCTGGAGCTACAGAGCCCAGGAGAGAGACCAGAGAGATCCTGCCCCACCCACCCACCC ACGCACTGTCCTCTCCCCATGCTAGCGCTTCACGGATGGCATCACCAACAAGCTGGTGG CCTGCTATGTGGAGGAGGACATGCAGGACTGCGTGCTGGTCCGGGTGTATGGGGAGCGG ACGGAGCTGCTGGTGGACCGGGAGAATGAGGTCAGAAACTTCCAGCTGCTGCGAGCACA CAGCTGTGCCCCCAAACTCTACTGCACCTTCCAGAATGGGCTGTGCTATGAGTACATGC AGGGTGTGGCCCTGGAGCCTGAGCACATCCGTGAGCCCCGGCTTTTCAGGTGAGGAGGG TGCCAGGGCCTCTGTCTCTACTATTCTTCAGGCCTTGGGCTTGGAAATCCTTGGCATGT GGGGTCCATGGTGGAGAGTTGTAAGATCTGTTGCCCTGGTGGGCCTGGAGGGTCCCTAT CTGGCCAGGAGGAAATGAGTGGGGAAGGCAATGTCCCAGCACTGATGCTCTACTCTTCC TGTTCCCTGCTGTAGGTTAATCGCCTTAGAAATGGCAAAGATTCATACTATCCACGCCA ACGGCAGCCTGCCCAAGCCCATCCTCTGGCACAAGATGCACAATTATTTCACGCTTGTG AAGAACGAGATCAACCCCAGGTACAAAGATCTGGGAGGGTCCAAGGCTGCCTTAATATG ATCTCCCTTCCCCATCCTTTTGGCTCCAGCCCACCCTGCTAGGAGGTTTAAGGGCTTAC AGTAAGACAGAGGGAGTGATGAGGCTCAGATTCAGGGCTCTTGAGGCCAAGCACCTGTC TATCAGCCCTCCTTTCACTGGCTACCTTTTTTGCCACTGCCCAGCCTTTCTGCAGATGT CCCTAAGGTAGAGGTGTTGGAACGGGAGCTGGCCTGGCTGAAGGAGCATCTGTCCCAGC TGGAGTCCCCTGTGGTGTTTTGTCACAATGACCTGCTCTGCAAGAATATCATCTATGAC AGCATCAAAGGTATGGCTTCTCTGGCCCTGGGGCAGCAGCAGGGTTTTGGTTGGTTGGT TAGCTGGCTTCACGGAGCTGTTCTTCCGGCTGGCACTTGGACAAGCAAAAGTGCATCAC TCAGTGGGTGCATCCCAGAGGTCTGTGGAAGCTTGACCAGGCTCTGTTGCCAGATCTCC TGGGCCAACTCTCATCGATTCTCCTCTACTTCATAGGTCACGTGCGGTTCATTGACTAT GAATATGCTGGCTACAACTACCAAGCTTTTGACATTGGCAACCATTTCAATGAGTTTGC AGGTGAGAGGGGCATTTCTACAGTCATGTGCTTTGTAGCGGTGTTTCAGTCAATGACTG ACCGCATGTAGATGGTGGTCCTATGAGATTACTGTATATTTACGAAGGAGATGGGAGTT TGAGGGGAGATGGGAACACCTACTGAGCTTGAGGAGACCCTTGGTGGGCCCATCTCTGA GCAACTTTGTCTGGCCTGCCTCCCCCTCAGGCGTGAATGAGGTGGATTACTGCCTGTAC CCGGCGCGGGAGACCCAGCTGCAGTGGCTGCACTACTACCTGCAGGCACAAAAGGGGAT GGCCGTGACCCCCAGGGAGGTGCAAAGGCTCTACGTGCAAGTCAACAAGTTTGCCCTGG TGAGTGCCTTATTTCTAGGGCTGGGGCAGGAAAGGAGGAGTTAGCAGGAGGGACTGGTG ACCCACCTAGCCTCCTACCCAGAGCCCAGGGTCAGGTGAGAGCCTGGGGACACCCATCC CTGGTTTCCCCAGCTTGTGCTAGGGAGACCCCACCACATCTCTTCCAGGGTCCTAGCTG TGTGTCTTCCACAATGACTGCATCCCTCCAGTGCTGTAGAGTCGGAAACAGGCATGGGG AGATTGCCAGGCTGACCCTCTCTGGTCTGTTTCCAGGCGTCTCACTTCTTCTGGGCTCT CTGGGCCCTCATCCAGAACCAGTACTCCACCATCGACTTTGATTTCCTCAGGTGAGTGT GGGGGTGATGGTGGGACGTGGAGAAAAGGGAGAGCCTTGGTTGAGCCCAGGGTGTGGGC AAAGGACAGCCCAGGCAGAATGCCCAGAGTCCGAGGCCTAGAGCCACTGAAAAACTTTT CTCATGATGTCAGCACCCCCAGCAGGGCCCACCTGGCTTTCTTCCTCACACCCAGCTTC CTTCTGTCTTCCCCACAGGTACGCAGTGATCCGATTCAACCAGTACTTCAAGGTGAAGC CTCAAGCGTCAGCCTTGGAGATGCCAAAGTGACCAGCCACCCCATCCCTCCCCTACCCA TCTGTCTGGCCAGACCTGTTCTCCAGAGCTCAATTCTGCACTCTGGGATCCACACCCTT GGACAGGGTG
If you do pairwise comparisons between the two protein sequences on one hand, and between the two corresponding cDNAs on the other (you can use lalign), you will notice a discrepancy. Looking at the exon coordinates in the GeneWise output, can you resolve it?
Tricks: Use the GeneWise web server at http://www.ebi.ac.uk/Wise2/. To speed up this task, you are provided with a trimmed version of the genomic human region, which corresponds to all the BLAST HSPs against either protein plus 100 bp on either side of the HSPs. Make sure you select the output option cDNA in the GeneWise form, so that you can retrieve the predicted cDNA for both splice variants! You are advised to copy the full GeneWise output into a text document for further reference.
Practice 4.5: Predicting splice variants in the mouse
The mouse orthologous locus of the human gene studied above, is given here (untrimmed): MOUSE_GENOMIC_1 (about 17 kb). This mouse genomic region region has not been investigated yet.
You are asked to map the two human protein sequences from above to it. Again, select the cDNA and translation output options in GeneWise. Looking at the results, which of the two human protein sequences do you trust more? (you can compare the mouse and human proteins using lalign as in the previous exercise).
Tricks: Each of these analyses will run for about 3 min on the GeneWise server, so maybe you can do these exercises in larger groups together.
5.- Managing Other Databases
Practice 5.1: Metabolic / Enzyme databases
- Go to the Genome Net server (Japan)
- Find a database called KEGG
- In KEGG, find the enzyme number EC 1.2.3.4
- Have a look at BRENDA database from the KEGG entry.
- Get from KEGG the ENZYME entry in ExPASy, then from ENZYME the UniProtKB/Swiss-Prot entry.
- Using Brenda database: find all enzymes using FAD as cofactor.
- At KEGG pathway: look for the Krebs cycle: compare this pathway in E.coliK12 and in human.
Tricks: [solution]
Practice 5.2: Polymorphisms
- Look for information in dbSNP on the human blue eye variant rs12913832
- What is the Craig Venter's 'eye color' (Celera genome assembly) ?
- Follow the link to the Alfred database to look for the population distribution of the 'blue eye allele'.
Tricks: [solution]
Practice 5.3: Publication databases
- How many papers did Nature publish in 1995 ?
- Find the publication dealing with Dolly death and find its DOI number.
- Get the publication thank to this DOI number on the site http://dx.doi.org
- How many articles are dealing with Viagra ? What is the 'generic' name of the molecule ?
Tricks: [solution]
6.- Tools: Pairwise Alignments
Practice 6.1: Compare the sequences OPRM_RAT and SSR1_HUMAN (these are the SWISS-PROT IDs) with lalign using default parameters.
The sequences can be fetched
here (choose the "FASTA" format) using the SWISS-PROT IDs.
Don't hesitate to look at the complete SWISS-PROT entries (OPRM_RAT
and SSR1_HUMAN),
in order to get more information about these two proteins !
Try to answer the following questions:
- Is this a local or global alignment ?
- Switch between local and global alignment . Try to understand the differences.
- Why are there several alignments displayed when performing the local alignment ?
- What does "% identity" mean ? How is it computed ?
- What do the symbols ":" and "." stand for ?
- When two residues are different, there can be either a "." or a blank. Try to understand the difference and what parameters influence this result ?
- Try to modify the gap penalties, examine more closely how these parameters influence the occurrence and the length of gaps ("-").
- Try to modify the scoring matrices used (i.e. BLOSUM35 and BLOSUM80), examine more closely how these parameters influence the scores and the alignments.
Tricks: see the following hints
Practice 6.2: Dotplot using Dotlet
Compare the same sequences (OPRM_RAT and SSR1_HUMAN) using Dotlet.
The sequences can be fetched here (choose the "FASTA" format).
Start with a look onto the Dotlet documentation
- Load the two sequences into Dotlet and compute the dotplot.
- What does the intensity (gray level) of a pixel mean ?
- Try changing the grayscale borders. Where would be an optimal position for the upper and lower limits of the grayscale ?
- What do the diagonal lines represent ?
- Try to identify corresponding aligned regions in the dotplot and the alignments found by LALIGN.
- Try to modify the noise by changing the window size, the threshold, both.
- Try comparing each sequence against itself.
Tricks: Dotlet examples and method comparison. The Dotlet learn by example pages show different typical sequence analysis problems. Take an interested look at the Dotlet examples and try to understand the dotplots.
Practice 6.3: Perform a pairwise sequence alignment for the following analogous proteins at EBI
>539057 MATAGKVIECKAAVAWEAGKPLSIEEVEVAPPHAMEVRVKILYTALCHTDVYFWEAKGQTPVFPRILGHE AGGIVESVGEGVTELVPGDHVLPVFTGECKDCAHCKSEESNLCDLLRINVDRGVMIGDGQSRFTINGKPI FHFVGTSTFSEYTVIHVGCLAKINPEAPLDKVCVLSCGISTGLGATLNVAKPKKGSTVAIFGLGAVGLAA MEGARMAGASRIIGVDLNPAKYEQAKKFGCTDFVNPKDHTKPVQEVLVEMTNGGVDRAVECTGHIDAMIA AFECVHDGWGVAVLVGVPHKEAVFKTYPMNFLNERTLKGTFFGNYKPRTDLPEVVEMYMRKELELEKFIT HSVPFSQINTAFDLMLKGEGLRCIMRMDQ
>82347 MATAGKVIKCKAAVAWEAGKPLTMEEVEVAPPQAMEVRVKILFTSLCHTDVYFWEAKGQIPMFPRIFGHE AGGIVESVGEGVTDVAPGDHVLPVFTGECKECPHCKSAESNMCDLLRINTDRGVMIGDGKSRFSIGGKPI YHFVGTSTFSEYTVMHVGCVAKINPEAPLDKVCVLSCGISTGLGASINVAKPPKGSTVAIFGLGAVGLAA AEGARIAGASRIIGVDLNAVRFEEARKFGCTEFVNPKDHTKPVQQVLADMTNGGVDRSVECTGNVNAMIQ AFECVHDGWGVAVLVGVPHKDAEFKTHPMNFLNERTLKGTFFGNFKPRTDLPNVVEMYMKKELEVEKFIT HSVPFSEINTAFDLMAKGEGIRCIIRMDN
Practice 6.4: Perform a pairwise global alignment. [solution]
Download the P57727 protein sequence and compare with the following sequence
>splice variant MCSDDWKGHYANVACAQLGFPSYVSSDNLRVSSLEGQFREEFVSIDHLLPDDKVTALHHS VYVREGCASGHVVTLQCTACGHRRGYSSRIVGGNMSLLSQWPWQASLQFQGYHLCGGSVI TPLWIITAAHCVYDLYLPKSWTIQVGLVSLLDNPAPSHLVEKIVYHSKYKPKRLGNDIAL MKLAGPLTFNEMIQPVCLPNSEENFPDGKVCWTSGWGATEDGAGDASPVLNHAAVPLISN KICNHRDVYGGIISPSMLCAGYLTGGVDSCQGDSGGPLVCQERRLWKLVGATSFGIGCAE VNKPGVYTRVTSFLDWIHEQMERDLKT
Compare the results you obtained with the results obtained by doing a pairwise local alignment between the same pairs of sequences. How can you explain the differences? Which domain is missing in the splice variant of the protein?
Tricks: Use Align tools at EBI.
7.- Multiple Sequence Alignment
Practice 7.1: Generate a multiple Sequence Alignment of the botulinum toxins types A to G found in former exercises (Managing protein databases, practice 5). Use CLUSTALW in server "proteo.ibmc.umh.es".
Tricks: Use ssh to connect to the server. Make a directory and copy the fasta formated sequences. Invoke the program by typing "clustalw" and following the instructions. If you get "command not found", type "locate clustalw" and follow the absolute path.
Practice 7.2: Orthology and Paralogy. Go to the MyHits web site (myhits.isb-sib.ch), select the PROTEIN Hub from the Hub menu, paste these 3 SwissProt ID or AC: [solution]
sw:STMN2_HUMAN Q93045
sw:STM2A_XENLA Q09001
sw:STMN1_MOUSE P54227
Send the proteins to either ClustalW or T-Coffee to produce the multiple alignment. Send the resulting alignment to the MSA Hub and start the Jalview applet. From the Calculate menu of the applet, create a Neighbour Joining Tree.
- Discuss the results. What is wrong? Propose a strategy.
- Add sequences from SwissProt in your list, make a new multiple
sequence alignment and a tree as before (make sure to include the
STMN2 sequences from rat, human and mouse)
- Discuss the results and propose a new strategy in order to obtain a better tree.
Tricks: Make a multiple sequence alignment with CLUSTALW and set the output to phylip format. [hints]
Make a MSA with the DNA and restrict the coding regions. To compute the tree, use infobiogene. To plot a tree, you may use phylodendron or TreeView.
Remember: Never use a clustalw guide tree file (.dnd) in place of a phylogenetic tree.
Practice 7.3: Use the 4 sequences below and produce a multiple alignment: [solution]
>ens:ENSP00000354426 MAAGPSGCLVPAFGLRLLLATVLQAVSAFGAEFSSEACRELGFSSNLLCSSCDLLGQFNL LQLDPDCRGCCQEEAQFETKKLYAGAILEVCGUKLGRFPQVQAFVRSDKPKLFRGLQIKY VRGSDPVLKLLDDNGNIAEELSILKWNTDSVEEFLSEKLERI >ens:ENSRNOP00000036726 MAAGQGGWLRPALGLRLLLATAFQAVSALGAEFSSEACRELGFSSNLLCSSCDLLGQFNL LPLDPVCRGCCQEEAQFETKKLYAGAILEVCGUKLGRFPQVQAFVRSDKPKLFRGLQIKY VRGSDPVLKLLDDNGNIAEELSILKWNTDSVEEFLSEKLERI >ens:ENSMUSP00000046910 MAAGQGGWLRPALGLRLLLATAFQAASALGAEFASEACRELGFSSNLLCSSCDLLGQFNL LPLDPVCRGCCQEEAQFETKKLYAGAILEVCGUKLGRFPQVQAFVRSDKPKLFRGLQIKY VRGSDPVLKLLDDNGNIAEELSILKWNTDSVEEFLSEKLERI >ens:ENSANGP00000011457 MRLFAITCLLFSIVTVIGAEFSAEDCRELGLIKSQLFCSSCSSLSDYGLIELKEHCLECC QKDTEADSKLKVYPAAVLEVCTCKFGAYPQIQAFIKSDRPAKFPNLTIKYVRGLDPIVKL MDEQGNVKETLSINKWNTDTVQEFFETRLAKVEDDDYLKTNRV
Tricks: [hint]
Practice 7.4: Use BioEdit to identify the consensus sequence for the following somatostatins
gi_6678034 ref NM—09215.1 Mouse somatostatin (Smst), mRNA
AGCGGCTGAAGGAGACGCTACCGAAGCCGTCGCTGCTGCCTGAGGACCTGCGACTAGACTGACCCA CCGCGCTCCAGCTTGGCTGCCTGAGGCAAGGAAGATGCTGTCCTGCCGTCTCCAGTGCGCCCTGGC TGCGCTCTGCATCGTCCTGGCTTTGGGCGGTGTCACCGGCGCGCCCTCGGACCCCAGACTCCGTCA GTTTCTGCAGAAGTCTCTGGCGGCTGCCACCGGGAAACAGGAACTGGCCAAGTACTTCTTGGCAGA GCTGCTGTCCGAGCCCAACCAGACAGAGAATGATGCCCTGGAGCCCGAGGATTTGCCCCAGGCAGC TGAGCAGGACGAGATGAGGCTGGAGCTGCAGAGGTCTGCCAACTCGAACCCAGCAATGGCACCCCG GGAACGCAAAGCTGGCTGCAAGAACTTCTTCTGGAAGACATTCACATCCTGTTAGCTTTAATATTG TTGTCCTAGCCAGACCTCTGATCCCTCTCCCCCAAACCCCATATCTCTTCCTTAACTCCTGGCCCC CGATGCTCAACTTGACCCTGCATTAGAAATTGAAGACTGTAAATACAAAATAAAATTATGGTGAGA TTATG
gi_207030_gb_M25890.1_RATSOMX Rat somatostatin mRNA, complete cds
TGCGGACCTGCGTCTAGACTGACCCACCGCGCTCAAGCTCGGCTGTCTGAGGCAGGGGAGATGCTG TCCTGCCGTCTCCAGTGCGCGCTGGCCGCGCTCTGCATCGTCCTGGCTTTGGGCGGTGTCACCGGG GCGCCCTCGGACCCCAGACTCCGTCAGTTTCTGCAGAAGTCTCTGGCGGCTGCCACCGGGAAACAG GAACTGGCCAAGTACTTCTTGGCAGAACTGCTGTCTGAGCCCAACCAGACAGAGAACGATGCCCTG GAGCCTGAGGATTTGCCCCAGGCAGCTGAGCAGGACGAGATGAGGCTGGAGCTGCAGAGGTCTGCC AACTCGAACCCAGCCATGGCACCCCGGGAACGCAAAGCTGGCTGCAAGAACTTCTTCTGGAAGACA TTCACATCCTGTTAGCTTTAATATTGTTGTCTCAGCCAGACCTCTGATCCCTCTCCTCCAAATCCC ATATCTCTTCCTTAACTCCCAGCCCCCCCCCCAATGCTCAACTAGACCCTGCGTTAGAAATTGAAG ACTGTAAATACAAAATAAAATTATGGTGAAATTATG
_gi_163636_gb_M31217.1_BOVPSOMA Bovine somatostatin mRNA, complete cds
AAGCTGCTTTAGGAGAGGCAAGGTTCGAGCCGTCGCTGCTGCCTGCGATCAGCTCCTAGAGTTTGA ACTCTAGCTCGGCTTCGCCGCCGCCGCCGAGATGCTGTCCTGCCGCCTCCAGTGCGCGCTGGCCGC GCTCTCCATCGTCCTGGCTCTTGGCGGTGTCACCGGCGCGCCCTCGGATCCCCGGCTCCGTCAGTT TCTGCAGAAATCCCTGGCTGCTGCCGCTGGCAAGCAGGAACTGGCCAAGTACTTCTTGGCAGAGCT GCTGTCTGAACCCAACCAGACAGAGATTGATGCCCTGGAGCCTGAAGATTTGTCCCAGGCTGCTGA GCAGGATGAAATGAGGCTGGAGCTGCAGAGATCTGCTAACTCAAACCCGGCCATGGCACCCCGAGA ACGCAAAGCTGGCTGCAAGAATTTCTTCTGGAAGACTTTCACATCCTGTTAACTTTATTAATGATT GTTGCCCATATAAGACCTCTGATTCCTCTTCTCCAAACCCCTTCTCACCTCCCTAATCCCTCCAAT CCTCAATAAGACCCTCGTGTTAGAAATTGAAGACTGTAAATACAAAATAAAATTATGGGAAATTAT G
Tricks: Install BioEdit in your computer and run it. Create a new alignment (File/New Alignment). Then add sequences and names in Sequences/New. You will see the sequences in italic and black and white. Save file (File/Save as...) and reload the file. Now you will see the sequences in full colour and most functions activated. Make an MSA in BioEdit (Accessory Applications/CLUSTALW), and then Alignment/Create consensus sequence. Colour the alignment to highlight the common areas.
Practice 7.5: Make a MSA of the following sequences to align correctly the active center of the catalytic domain of kinases.
>D28 CD28 S. CEREVISIAE CELL CYCLE CONTROL PROTEIN KINASE ANYKRLEKVGEGTYGVVYKALDLRPGQGQRVVALKKIRLESEDEGVPSTAIREISLLKEL KDDNIVRLYDIVHSDAHKLYLVFEFLDLDLKRYMEGIPKDQPLGADIVKKFMMQLCKGIA YCHSHRILHRDLKPQNLLINKDGNLKLGDFGLARAFGVPLRAYTHEIVTLWYRAPEVLLG GKQYSTGVDTWSIGCIFAEMCNRKPIFSGDSEIDQIFKIFRVLGTPNEAIWPDIVYLPDF KPSFPQWRRKDLSQVVPSLDPRGIDLLDKLLAYDPINRISARRAAIHPYFQES >SKH SKH HELA MYSTERY PUTATIVE PROTEIN KINASE AKYDIKALIGRGSFSRVVRVEHRATRQPYAIKMIETKYREGREVCESELRVLRRVRHANI IQLVEVFETQERVYMVMELATGGELFDRIIAKGSFTERDATRVLQMVLDGVRYLHALGIT HRDLKPENLLYYHPGTDSKIIITDFGLASARKKGDDCLMKTTCGTPEYIAPEVLVRKPYT NSVDMWALGVIAYILLSGTMPFEDDNRTRLYRQILRGKYSYSGEPWPSVSNLAKDFIDRL LTVDPGARMTALQALRHPWVVSM >APK CAPK BOVINE CARDIAC MUSCLE CYCLIC AMP-DEPENDENT (ALPHA) DQFERIKTLGTGSFGRVMLVKHMETGNHYAMKILDKQKVVKLKQIEHTLNEKRILQAVNF PFLVKLEFSFKDNSNLYMVMEYVPGGEMFSHLRRIGRFSEPHARFYAAQIVLTFEYLHSL DLIYRDLKPENLLIDQQGYIQVTDFGFAKRVKGRTWTLCGTPEYLAPEIILSKGYNKAVD WWALGVLIYEMAAGYPPFFADQPIQIYEKIVSGKVRFPSHFSSDLKDLLRNLLQVDLTKR FGNLKDGVNDIKNHK >EE1 WEE1 S. POMBE MITOTIC INHIBITOR TRFRNVTLLGSGEFSEVFQVEDPVEKTLKYAVKKLKVKFSGPKERNRLLQEVSIQRALKG HDHIVELMDSWEHGGFLYMQVELCENGSLDRFLEEQGQLSRLDEFRVWKILVEVALGLQF IHHKNYVHLDLKPANVMITFEGTLKIGDFGMASVWPVPRGMEREGDCEYIAPEVLANHLY DKPADIFSLGITVFEAAANIVLPDNGQSWQKLRSGDLSDAPRLSSTDNGSSLTSSSRETP ANSIIGQGGLDRVVEWMLSPEPRNRPTIDQILATDEVCWV >GFR EGFR HUMAN EPIDERMAL GROWTH FACTOR RECEPTOR TEFKKIKVLGSGAFGTVYKGLWIPEGEKVKIPVAIKELREATSPKANKEILDEAYVMASV DNPHVCRLLGICLTSTVQLITQLMPFGCLLDYVREHKDNIGSQYLLNWCVQIAKGMNYLE DRRLVHRDLAARNVLVKTPQHVKITDFGLAKLLGAEEKEYHAEGGKVPIKWMALESILHR IYTHQSDVWSYGVTVWELMTFGSKPYDGIPASEISSILEKGERLPQPPICTIDVYMIMVK CWMIDADSRPKFRELIIEFSKMAR >DGM PDGF RECEPTOR, MOUSE KINASE REGION DQLVLGRTLGSGAFGQVVEATAHGLSHSQATMKVAVKMLKSTARSSEKQALMSELYGDLV DYLHRNKHTFLQRHSNKHCPPSAELYSNALPVGFSLPSHLNLTGESDGGYMDMSKDESID YVPMLDMKGDIKYADIESPSYMAPYDNYVPSAPERTYRATLINDSPVLSYTDLVGFSYQV ANGMDFLASKNCVHRDLAARNVLICEGKLVKICDFGLARDIMRDSNYISKGSTYLPLKWM APESIFNSLYTTLSDVWSFGILLWEIFTLGGTPYPELPMNDQFYNAIKRGYRMAQPAHAS DEIYEIMQKCWEEKFETRPPFSQLVLLLERLLGEGYKKKY >FES THIS IS VFES TYROSINE KINASE VLNRAVPKDKWVLNHEDLVLGEQIGRGNFGEVFSGRLRADNTLVAVKSCRETLPPDIKAK FLQEAKILKQYSHPNIVRLIGVCTQKQPIYIVMELVQGGDFLTFLRTEGARLRMKTLLQM VGDAAAGMEYLESKCCIHRDLAARNCLVTEKNVLKISDFGMSREAADGIYAASGGLRQVP VKWTAPEALNYGRYSSESDVWSFGILLWETFSLGASPYPNLSNQQTREFVEKGGRLPCPE LCPDAVFRLMEQCWAYEPGQRPSFSAIYQEL >AF1 RAF1 HUMAN C-RAF-1 ONCOGENE SEVMLSTRIGSGSFGTVYKGKWHGDVAVKILKVVDPTPEQFQAFRNEVAVLRKTRHVNIL LFMGYMTKDNLAIVTQWCEGSSLYKHLHVQETKFQMFQLIDIARQTAQGMDYLHAKNIIH RDMKSNNIFLHEGLTVKIGDFGLATVKSRWSGSQQVEQPTGSVLWMAPEVIRMQDNNPFS FQSDVYSYGIVLYELMTGELPYSRDQIIFMVGRGYASPDLSKLYKNCPKAMKRLVADCVK KVKEERPLFPQILSSIELLQH >MOS CMOS HUMAN C-MOS ONCOGENE EQVCLLQRLGAGGFGSVYKATYRGVPVAIKQVNKCTKNRLASRRSFWAELNVARLRHDNI VRVVAASTRTPAGSNSLGTIIMEFGGNVTLHQVIYGAAGHPEGDAGEPHCRTGGQLSLGK CLKYSLDVVNGLLFLHSQSIVHLDLKPANILISEQDVCKISDFGCSEKLEDLLCFQTPSY PLGGTYTHRAPELLKGEGVTPKADIYSFAITLWQMTTKQAPYSGERQHILYAVVAYDLRP SLSAAVFEDSLPGQRLGDVIQRCWRPSAAQRPSARLLLVDLTSLKA >SVK HSVK HERPES SIMPLEX VIRUS PUTATIVE PROTEIN KINASE MGFTIHGALTPGSEGCVFDSSHPDYPQRVIVKAGWYTSTSHEARLLRRLDHPAILPLLDL HVVSGVTCLVLPKYQADLYTYLSRRLNPLGRPQIAAVSRQLLSAVDYIHRQGIIHRDIKT ENIFINTPEDICLGDFGAACFVQGSRSSPFPYGIAGTIDTNAPEVLAGDPYTTTVDIWSA GLVIFETAVHNASLFSAPRGPKRGPCDSQITRIIRQAQVHVDEFSPHPESRLTSRYRSRA AGNNRPPYTRPAWTRYYKMDIDV
Tricks: Evaluate the performance of the four types of alignments: CLUSTALW, MAP, PIMA and BlockMaker in the BCM server. To simplificate the evaluation, classificate the alignment type as "all or none", whether all sequences have been correctly aligned in the server, or not.
8.- Similarity Searches: BLAST and PSI-BLAST
Practice 8.1: Make a similarity search with the protein insulin from D. rerio
>gi|12053668|emb|CAC20109.1|insulin[Danio rerio] MAVWIQAGALLVLLVVSSVSTNPGTPQHLCGSHLVDALYLVC GPTGFFYNPKRDVEPLLGFLPPKSAQETEVADFAFKDHAELIR KRGIVEQCCHKPCSIFELQNYCN
Tricks: Start at the NCBI homepage:
http://www.ncbi.nlm.nih.gov/.
Click on ‘BLAST’ at the top menu.
This is the starting point for several BLAST programs available from the
BLAST homepage. Since insulin is a protein sequence, we will search
using BLASTP.
Click on ‘Protein-protein BLAST [blastp]’ under the Protein BLAST
heading. The BLASTp page appears. Contains a query box as well as
several optional parameters that can be set. Copy and past a query sequence into the query box. The remainder of the search form allows you to set various options. Set
subsequence - allows you to search with a particular portion of the
sequence. Leave it blank so that the entire sequence will be used in the
search. Choose database has a drop-down menu that allows you to choose
which part of the database you want to search. Leave it on ‘nr’ (for
non-redundant), for the complete database.
Do CD-Search allows a comparison of the query sequence to a database of
conserved domain patterns. This is a powerful tool for finding
functional domains in genes. Leave it toggled on.
Click BLAST! to start search.
First shown is the result of the CD-Search (Conserved Domain search).
Click on the image to see the full list of conserved domains. A pop up
window appears. Two known domain patterns are identified: insulin and
insulin-growth factor. Roll over the bars. The text in the above box
should change. ‘S’ (the alignment score) indicates how strong the match
was. Higher is better. ‘E’ (the expect value) is a statistical measure
of the significance of the match. It is the expectation that the match
be found in the database by chance alone. Lower is better.
• Close the pop up window.
• Click FORMAT! to send the results of the actual search Components of
Output:
1. Graphical display of the strongest matches to the query sequence.
Here, some hits are to full-length insulin and some are to the shorter
processed form. Color coded according to the alignment score. Roll over
the various lines. The identification information, alignment score (S)
and E value appear in the text box above the chart.
2. Detailed list of hits ordered by their alignment scores. They
correspond to the ones displayed graphically. Entries are ranked from
the lowest to the highest E value (i.e. most similar to more distant)
3. Actual gene alignments. Empty spaces indicate mismatches. + indicates
similarity between the two different amino acids compared. A gap (XXXX)
is inserted to give a good alignment with the query. This would be a
site of an insertion or deletion even during evolution.
Practice 8.2: Human dystrophyn similarity searches. Make a blast using the dystrophyn accession number
How many types of conserved
domains are shown? how many colors are shown? What are their full
names?
Click on the score (1930) for the mouse gene (gi|192972|gb|AAA37530.1|
dystrophin) Are these sequences similar to that of the human query
sequence? Has there been divergence between the two sequences? How do
you know?
Given longer periods of evolutionary separation, what might you expect
to see? Now let’s ask whether Drosophila has a dystrophin sequence. Make
a new blast search against Drosophila melanogaster.
Has the Drosophila dystrophin gene product diverged from the human
version? How do you know this? If you were to examine the two sequences
more carefully, what might you expect?
Based on your answer to the last question, do you think human is that
different from the fly?
Tricks: Go to NCBI-Blast. Type the accession number ‘P11532’ in the query box, and press BLAST!. See "conserved domains". These small patterns identify the amino acid residues that are almost always conserved through evolution in particular functional domains. For the second blast yype P11532 again into the query box. Scroll down to Options for advanced blasting, select ‘Drosophila melanogaster’ from the ‘select from’ drop-down menu, and click BLAST!. Click on the top ranked score.
Practice 8.3: Submit the following nucleotide sequence to homology search with the BLAST tool at one of the three major nucleotide sequence databases and identify it
CCCAGCGCACCCGCACCATGGCCGGCCCCAGCCTCGCTTGCTGTCTGCTCGGCCTCCTGGCGCTGA CCTCCGCCTGCTACATCCAGAACTGCCCCCTGGGAGGCAAGAGGGCCGCGCCGGACCTCGACGTGC GCAAGTGCCTCCCCTGCGGCCCCGGGGGCAAAGGCCGCTGCTTCGGGCCCAATATCTGCTGCGCGG AAGAGCTGGGCTGCTTCGTGGGCACCGCCGAAGCGCTGCGCTGCCAGGAGGAGAACTACCTGCCGT CGCCCTGCCAGTCCGGCCAGAAGGCGTGCGGGAGCGGGGGCCGCTGCGCGGTCTTGGGCCTCTGCT GCAGCCCGGACGGCTGCCACGCCGACCCTGCCTGCGACGCGGAAGCCACCTTCTCCCAGCGCTGAA ACTTGATGGCTCCGAACACCCTCGAAGCGCGCCACTCGCTTCCCCCATAGCCACCCCAGAAATGGT GAAAATAAAATAAAGCAGGTTTTTCTCCTCT
Tricks: Use WU-BLAST2 Nucleotide at EBI or NCBI
Practice 8.4:
Perform a similarity search of a protein with the
following amino acid sequence.
Which protein is? Which specie does belong to?
Tricks: Use WU-BLAST2 Protein at
EBI or at
NCBI
Practice 8.5: Database search with Blastp. Use the protein sequence CDC16_SCHPO as query to search
for similar sequences in the Swiss-Prot database, using the
program
blastp. Use the default parameters and try to answer the
following questions: Tricks: Use basic BLAST at
EmbNet Practice 8.6: Database search with Blastx The EMBL W99073 sequence is a mouse EST (Warning:
ESTs are DNA sequences!). Compare this nucleotide sequence to the
Swiss-Prot database using the blastx program on
the
basic blast page. Tricks: Use basic BLAST at
EmbNet
Practice 8.7: Do a PSI-BLAST using the previous alignment (Multiple
Sequence Alignment, Practice 3) searching
against SwissProt+TrEMBL+ENSEMBL with both inclusion and report
thresholds set to 1-e6 Tricks: Practice 8.8: Given two sequences (P40623
and P26586), and assuming that P26586 is completely unknown, compare
the sequences and make a functional model based in homology to
predict the function of P26586. This model should indicate which
residues are homologues between the sequences. Tricks: Compare the sequences with
DOTTER or DOTTLET. Extract some fragments and look for alignments (be
generous in extracting fragments; later will be discarded unuseful
fragments). Check visually the alignments, specially in low
complexity regions. This could be a clue for fragment selection. To
improve alignments, add and "extra" fragment and compare pairwise or
MS alignment. Use BLAST to get close homologues. Use tools in Swiss-Prot
and PROSITE to guess the function.>gi_67428_pir
RTDCYGNVNRIDTTGASCKTAKPEGLSYCGVSASKKIAERDLQAMDRYKTIIKKVGEKLCVEPAVIAGII
SRESHAGKVLKNGWGDRGNGFGLMQVDKRSHKPQGTWNGEVHITQGTTILINFIKTIQKKFPSWTKDQQL
KGGISAYNAGAGNVRSYARMDIGTTHDDYANDVVARAQYYKQHGY
Try to answer the following questions:
Practice 8.9: Try to characterize the gene Xrcc1 for DNA reparing in humans. Answer the following questions: How this protein acts? Which domains? What it looks like?
Tricks: Use BLAST to find homologues. Compare the sequences using DOTTLET or DOTTER. Make a MSA of the homologue fragments. Use PSI-BLAST to find remote homologues with one of the fragments obtained before.
9.- Patterns, Profiles, and HMMs
Practice 9.1: PROSITE database
The aim of the exercise is to explore and understand the PROSITE database. First have a look at the following PROSITE entry: PS50235.
PS50235 is a Pattern or a Profile? How do you distinguish a Pattern from a Profile?
If this descriptor match your sequences (using standard parameters), will you believe the result? And if it doesn't match?
What is the function of PS50235? Is the PS50235 related to other PROSITE entries? If yes, are they Patterns or Profiles?
Tricks: Look at the Numerical results section. Check also the PDOC documentation
Practice 9.2: PROSITE vs. InterPro
Analyze the following sequence using InterProScan and ScanProsite:
>seq1 MELRVLLCWASLAAALEETLLNTKLETADLKWVTFPQVDGQWEELSGLDEEQHSVRTYEV CDVQRAPGQAHWLRTGWVPRRGAVHVYATLRFTMLECLSLPRAGRSCKETFTVFYYESDA DTATALTPAWMENPYIKVDTVAAEHLTRKRPGAEATGKVNVKTLRLGPLSKAGFYLAFQD QGACMALLSLHLFYKKCAQLTVNLTRFPETVPRELVVPVAGSCVVDAVPAPGPSPSLYCR EDGQWAEQPVTGCSCAPGFEAAEGNTKCRACAQGTFKPLSGEGSCQPCPANSHSNTIGSA VCQCRVGYFRARTDPRGAPCTTPPSAPRSVVSRLNGSSLHLEWSAPLESGGREDLTYALR CRECRPGGSCAPCGGDLTFDPGPRDLVEPWVVVRGLRPDFTYTFEVTALNGVSSLATGPV PFEPVNVTTDREVPPAVSDIRVTRSSPSSLSLAWAVPRAPSGAVLDYEVKYHEKGAEGPS SVRFLKTSENRAELRGLKRGASYLVQVRARSEAGYGPFGQEHHSQTQLDESEGWREQLAL IAGTAVVGVVLVLVVIVVAVLCLRKQSNGREAEYSDKHGQYLIGHGTKVYIDPFTYEDPN EAVREFAKEIDVSYVKIEEVIGAGEFGEVCRGRLKAPGKKESCVAIKTLKGGYTERQRRE FLSEASIMGQFEHPNIIRLEGVVTNSMPVMILTEFMENGALDSFLRLNDGQFTVIQLVGM LRGIASGMRYLAEMSYVHRDLAARNILVNSNLVCKVSDFGLSRFLEENSSDPTYTSSLGG KIPIRWTAPEAIAFRKFTSASDAWSYGIVMWEVMSFGERPYWDMSNQDVINAIEQDYRLP PPPDCPTSLHQLMLDCWQKDRNARPRFPQVVSALDKMIRNPASLKIVARENGGASHPLLD QRQPHYSAFGSVGEWLRAIKMGRYEESFAAAGFGSFELVSQISAEDLLRIGVTLAGHQKK ILASVQHMKSQAKPGTPGGTGGPAPQY
What is the domain composition of the protein? What is its function? Can you predict binding sites with InterPro? With PROSITE?
Tricks: Move the mouse on the sequences and images in the result page to highlight information.
Practice 9.3. Protein function discovery. [solution]
- Use different methods to find protein domains in the following sequence:
>mysterious sequence NNRNQDNYVSWSDSEDDDEDEEIEEKEKPETNFPSPFTNILCGIIFVERRYTAVVLNRLI KEAGKQDPELAYISSNFITGHGIGKNQPRNKQMEAEFRKQEEVLRKFRAHETNLLIATSI VEEGVDIPKCNLVVRFDLPTEYRSYVQSKGRARAPISNYIMLADTDKIKSFEEDLKTYKA IEKILRNKCSKSVDTGETDIDPVMDDDDVFPPYVLRPDDGGPRVTINTAIGHINRYCARL PSDPFTHLAPKCRTRELPDGTFYSTLYLPINSPLRASIVGPPMSCVRLAERVVALICCEK LHKIGELDDHLMPVGKETVKYEEELDLHDEEETSVPGRPGSTKRRQCYPKAIPECLRDSY PRPDQPCYLYVIGMVLTTPLPDELNFRRRKLYPPEDTTRCFGILTAKPIPQIPHFPVYTR SGEVTISIELKKSGFMLSLQMLELITRLHQYIFSHILRLEKPALEFKPTDADSAYCVLPL NVVNDSSTLDIDFKFMEDIEKSEARIGIPSTKYTKETPFVFKLEDYQDAVIIPRYRNFDQ PHRFYVADVYTDLTPLSKFPSPEYETFAEYYKTKYNLDLTNLNQPLLDVDHTSSRLNLLT PRHLNQKGKALPLSSAEKRKAKWESLQNKQILVPELCAIHPIPASLWRKAVCLPSILYRL HCLLTAEELRAQTASDAGVGVRSLPADFRYPNLDFGWKKSIDSKSFISISNSSSAENDNY CKHSTIVPENAAHQGANRTSSLENHDQMSVNCRTLLSESPGKLHVEVSADLTAINGLSYN QNLANGSYDLANRDFCQGNQLNYYKQEIPVQPTTSYSIQNLYSYENQPQPSDECTLLSNK YLDGNANKSTSDGSPVMAVMPGTTDTIQVLKGRMDSEQSPSIGYSSRTLGPNPGLILQAL TLSNASDGFNLERLEMLGDSFLKHAITTYLFCTYPDAHEGRLSYMRSKKVSNCNLYRLGK KKGLPSRMVVSIFDPPVNWLPPGYVVNQDKSNTDKWEKDEMTKDCMLANGKLDEDYEEED EEEESLMWRAPKEEADYEDDFLEYDQEHIRFIDNMLMGSGAFVKKISLSPFSTTDSAYEW KMPKKSSLGSMPFSSDFEDFDYSSWDAMCYLDPSKAVEEDDFVVGFWNPSEENCGVDTGK QSISYDLHTEQCIADKSIADCVEALLGCYLTSCGERAAQLFLCSLGLKVLPVIKRTDREK ALCPTRENFNSQQKNLSVSCAAASVASSRSSVLKDSEYGCLKIPPRCMFDHPDADKTLNH LISGFENFEKKINYRFKNKAYLLQAFTHASYHYNTITDCYQRLEFLGDAILDYLITKHLY EDPRQHSPGVLTDLRSALVNNTIFASLAVKYDYHKYFKAVSPELFHVIDDFVQFQLEKNE MQGMDSELRRSEEDEEKEEDIEVPKAMGDIFESLAGAIYMDSGMSLETVWQVYYPMMRPL IEKFSANVPRSPVRELLEMEPETAKFSPAERTYDGKVRVTVEVVGKGKFKGVGRSYRIAK SAAARRALRSLKANQPQVPNS
Tricks: InterPro is an integrated documentation resource for protein families, domains and sites. InterPro combines a number of databases (referred to as member databases) that use different methodologies and a varying degree of biological information on well-characterized proteins to derive protein signatures. By uniting the member databases, InterPro capitalizes on their individual strengths, producing a powerful integrated diagnostic tool. InterPro unifies:
- PROSITE regular expressions and profiles
- Pfam, SMART, TIGRFAMs, PIRSF, PANTHER, Gene3D and SUPERFAMILY hidden Markov models (HMMs)
- PRINTS, provider of fingerprints (groups of aligned, un-weighted motifs)
- PRODOM who use Clustr analysis to group sequences
Signatures describing the same protein family, domain repeat or site are grouped into unique InterPro entries. Each combined InterPro entry has a unique accession number, an abstract describing the features of proteins associated with the entry and literature references and has links to the relevant member database(s). All UniProtKB protein sequences that have matches to a particular InterPro entry are listed in the Match Table associated with that entry. There are also links to the InterPro graphical views. The graphical views, which can be sorted by UniProtKB accession number, structure or taxonomy, show the position of the signatures on the protein, mousing over the signature brings up a pop-box, giving the accession, name and position.
InterPro graphically represents the location of a protein domain and information pertaining to the origin of that domain and the proteins that contain it. Families are also defined and may contain several InterPro domains which are often, but not always, in the same order. Through the InterPro Domain Architecture view, the composition and order of the different domains within a family are clearly displayed for easy comparison, as well as for simple navigation between the entries for individual domains.
InterPro and InterProScan are accessible for interactive use over the EBI web server (www/ebi.ac.uk/interpro), they are distributed as stand-alone copies by anonymous ftp.
InterPro entries are linked to one another through PARENT/CHILD and CONTAINS/FOUND IN relationships. PARENT/CHILD relationships indicate superfamily/family/subfamily relationships, as well as domain hierarchies, where sequences can be subdivided into more specific sub-sets. CONTAINS/FOUND IN relationships apply to domains, repeats and sites within families, and are used to describe the composition of protein sequences.
Practice 9.4. PSI-BLAST. Given the following sequence: [solution]
>myseq MRTSSEDVFISDEVPTISPNTMLTSALMYMKDYITAVVVVDENRINVLVGMISTEDILDV KVGGFFKPAIVCNGDDTLDECRSAIPEGNAQHIPVLEGFDKFHGIIYDKDVLHFGPPPRR KPVAGGRGQAGAPGVLPKKRPGGPGSAGDDGAPPPPPSPKGVVGSKAAGGRGGTRGTGYA GEPAEHAGIAGGMRGPQPGPPTGPSGGPAGSGERGAKGDPGPPGQPSMAKKGTAGRESVT GILGSNGSGKSTFLNIMYGQDVFNEPTNGSNGWHNQDGQVIPMSVIGQKGVSPSLDVGDN LMEFNHVIERSGQLSKADYLGADSVLVMTEVKELYEELPNCLSGGWKQWVNLCLEMITKS
Questions:
- Can you give a possible function to the protein based on the homologies found?
- Can you distinguish some protein domains?
- What is particular to our protein in respect to its homologous?
- What does it append when you run the PSI-BLAST search without the low complexity filter?
Tricks: Go to the NCBI BLAST service and run a PSI-BLAST search using the myseq sequence against the SWISS-PROT database. Select the low complexity filter and keep the rest of the options as they are. Run a few rounds (at least 5) of PSI-BLAST.
Practice 9.5. Build a pattern. [solution]
- Given the following MSA:
Seq1 WFFKGIADKDAERHLLA Seq2 WFFKNLEQKDAEARLLA Seq3 WFFKR---KDAERQLLA Seq4 WFFGTI---DAERQLLA Seq5 WFFKDIPTKDAERQLLA Seq6 WYFG----RESERLLLA Seq7 WYFGKIPLKDAERQLLA Seq8 WYFGKLRAKDTERLLLL
build a pattern using a text editor.
- Submit the patten to the ScanProsite server and search against SWISS-PROT. Could you say something about the proteins matching your pattern?
- A possible way to validate the output of the pattern search is to search against a randomized database. To validate you pattern, use again ScanProsite, but this time against a reversed SWISS-PROT. Do you find any false positive sequence?
- Repeat the exercise with the following sequences:
seq1 ERGLR seq2 DRASR seq3 DRLGR seq4 ERAAR seq5 ERGVR
- What's append with a random database
Tricks: Use the Prosite syntax (you can find the syntax rules here).
Practice 9.6. Search the Prosite pattern database. [solution]
- Scan the human protein VAV_HUMAN against Prosite patterns using ScanProsite. Have a look to the results.
- Repeat the same scan but including patterns with a high probability of occurrence. What's the difference between this and the previous result? Which are the characteristics of the masked patterns?
Tricks:
Practice 9.7. Build PSSMs with MEME. [solution]
- Go to the MEME motif discovery service.
- Paste the following sequences (FASTA format) in the 'sequences
box' and select the option Any number of repetitions.
>seq1 MGFSSALQSRAAHEALIVRQDAELRLMETMKRSIQMKAKCDKEYAISLTAVAQQGLKIDRADEMQGSLISKSWRSYMDEL DHQAKQFKFNAEQLEVVCDKLTHLSQDKRKARKAYQEEHAKIAARLNHLTDEVVRKKSEYQKHLEGYKALRTRFEENYIK APSRSGRKLDDVRDKYQKACRKLHLTHNEYVLSITEAIEVEKDFRNVLLPGLLEHQQSVQESFILLWRNILQEAAQYGDL TADKYKEIQKRIDTVIGSINPTEEYGEFTEKYKTSPTTPLLFQFDETLIQDIPGKLQSSTLTVDNLTVDWLRNRLQELEG AVRDCQEKQMKMIEHVNGGSPVANGSIISNGSNTSNGIQSNKDSLCRQSKDLNALRCQEKQKQKLVDMIKCALNEVGCEE LPSGCDDDLTLEQNFIENGYNNEQQISLSTNRPLYEEEWFHGVLPREEVVRLLNNDGDFLVRETIRNEESQIVLSVCWNG HKHFIVQTTGEGNFRFEGPPFASIQELIMHQYHSELPVTVKSGAILRRPVCRERWELSNDDVVLLERIGRGNFGDVYKAK LKSTKLDVAVKTCRMTLPDEQKRKFLQEGRILKQYDHPNIVKLIGICVQKQPIMIVMELVLGGSLLTYLRKNSNGLTTRQ QMGMCRDAAAGMRYLESKNCIHRDLAARNCLVDLEHSVKISDFGMSREEEEYIVSDGMKQIPVKWTAPEALNFGKYTSLC DVWSYGILMWEIFSKGDTPYSGMTNSRARERIDTGYRMPTPKSTPEEMYRLMLQCWAADAESRPHFDEIYNVVDALILRL DNSH >seq2 MEAIAKYDFKATADDELSFKRGDILKVLNEECDQNWYKAELNGKGGFIPKNYIEMKPHPWFFGKIPRAKAEEMLGKQRHD GAFLIRESESAPGDFSLSVKFGNDVQQFKVLRDGAGKYLLWVVKFNSLNELVDYHRSTSVSRNQQIFLRDIEQVPQQPTY VQALFDFDPQEEGELGFRRGDFIQVLDNSDPNWWKGACHGQTGMFPRNYVTPVNRNI >seq3 MSAEGYQYRALYDYKKEREEDIDLHLGDILTVNKGSLVALGFSDGQEARPEEIGWLNGYNETTGERGDFPGTYVEYIGRK KISPPTPKPRPPRPLPVAPGSSKTEADVEQQALTLPDLAEQFAPPDIAPPLLIKLVEAIEKKGLECSTLYRTQSSSNLAE LRQLLDCDTPSVDLEMIDVHVLADAFKRYLLDLPNPVIPAAVYSEMISLAPEVQSSEEYIQLLKKLIRSPSIPHQYWLTL QYLLKHFFKLSQTSSKNLLNARVLSEIFSPMLFRFSAASSDNTENLIKVIEILISTEWNERQPAPALPPKPPKPTTVANN GMNNNMSLQNAEWYWGDISREEVNEKLRDTADGTFLVRDASTKMHGDYTLTLRKGGNNKLIKIFHRDGKYGFSDPLTFSS VVELINHYRNESLAQYNPKLDVKLLYPVSKYQQDQVVKEDNIEAVGKKLHEYNTQFQEKSREYDRLYEEYTRTSQEIQMK RTAIEAFNETIKIFEEQCQTQERYSKEYIEKFKREGNEKEIQRIMHNYDKLKSRISEIIDSRRRLEEDLKKQAAEYREID KRMNSIKPDLIQLRKTRDQYLMWLTQKGVRQKKLNEWLGNENTEDQYSLVEDDEDLPHHDEKTWNVGSSNRNKAENLLRG KRDGTFLVRESSKQGCYACSVVVDGEVKHCVINKTATGYGFAEPYNLYSSLKELVLHYQHTSLVQHNDSLNVTLAYPVYA QQRR >seq4 MDLLPPKPKYNPLRNESLSSLEEGASGSTPPEELPSPSASSLGPILPPLPGDDSPTTLCSFFPRMSNLRLANPAGGRPGS KGEPGRAADDGEGIDGAAMPESGPLPLLQDMNKLSGGGGRRTRVEGGQLGGEEWTRHGSFVNKPTRGWLHPNDKVMGPGV SYLVRYMGCVEVLQSMRALDFNTRTQVTREAISLVCEAVPGAKGATRRRKPCSRPLSSILGRSNLKFAGMPITLTVSTSS LNLMAADCKQIIANHHMQSISFASGGDPDTAEYVAYVAKDPVNQRACHILECPEGLAQDVISTIGQAFELRFKQYLRNPP KLVTPHDRMAGFDGSAWDEEEEEPPDHQYYNDFPGKEPPLGGVVDMRLREGAAPGAARPTAPNAQTPSHLGATLPVGQPV GGDPEVRKQMPPPPPCPGRELFDDPSYVNVQNLDKARQAVGGAGPPNPAINGSAPRDLFDMKPFEDALRVPPPPQSVSMA EQLRGEPWFHGKLSRREAEALLQLNGDFLVRESTTTPGQYVLTGLQSGQPKHLLLVDPEGVVRTKDHRFESVSHLISYHM DNHLPIISAGSELCLQQPVERKL >seq5 MAQWNQLQQLDTRYLEQLHQLYSDSFPMELRQFLAPWIESQDWAYAASKESHATLVFHNLLGEIDQQYSRFLQESNVLYQ HNLRRIKQFLQSRYLEKPMEIARIVARCLWEESRLLQTAATAAQQGGQANHPTAAVVTEKQQMLEQHLQDVRKRVQDLEQ KMKVVENLQDDFDFNYKTLKSQGDMQDLNGNNQSVTRQKMQQLEQMLTALDQMRRSIVSELAGLLSAMEYVQKTLTDEEL ADWKRRQQIACIGGPPNICLDRLENWITSLAESQLQTRQQIKKLEELQQKVSYKGDPIVQHRPMLEERIVELFRNLMKSA FVVERQPCMPMHPDRPLVIKTGVQFTTKVRLLVKFPELNYQLKIKVCIDKDSGDVAALRGSRKFNILGTNTKVMNMEESN NGSLSAEFKHLTLREQRCGNGGRANCDASLIVTEELHLITFETEVYHQGLKIDLETHSLPVVVISNICQMPNAWASILWY NMLTNNPKNVNFFTKPPIGTWDQVAEVLSWQFSSTTKRGLSIEQLTTLAEKLLGPGVNYSGCQITWAKFCKENMAGKGFS FWVWLDNIIDLVKKYILALWNEGYIMGFISKERERAILSTKPPGTFLLRFSESSKEGGVTFTWVEKDISGKTQIQSVEPY TKQQLNNMSFAEIIMGYKIMDATNILVSPLVYLYPDIPKEEAFGKYCRPESQEHPEADPGSAAPYLKTKFICVTPTTCSN TIDLPMSPRTLDSLMQFGNNGEGAEPSAGGQFESLTFDMDLTSECATSPM >seq6 MELWRQCTHWLIQCRVLPPSHRVTWEGAQVCELAQALRDGVLLCQLLNNLLPQAINLREVNLRPQMSQFLCLKNIRTFLS TCCEKFGLKRSELFEAFDLFDVQDFGKVIYTLSALSWTPIAQNKGIMPFPTEDSALNDEDIYSGLSDQIDDTAEEDEDLY DCVENEEAEGDEIYEDLMRLESVPTPPKMTEYDKRCCCLREIQQTEEKYTDTLGSIQQHFMKPLQRFLKPQDMETIFVNI EELFSVHTHFLKELKDALAGPGATTLYQVFIKYKERFLVYGRYCSQVESASKHLDQVATAREDVQMKLEECSQRANNGRF TLRDLLMVPMQRVLKYHLLLQELVKHTQDATEKENLRLALDAMRDLAQCVNEVKRDNETLRQITNFQLSIENLDQSLANY GRPKIDGELKITSVERRSKTDRYAFLLDKALLICKRRGDSYDLKASVNLHSFQVRDDSSGERDNKKWSHMFLLIEDQGAQ GYELFFKTRELKKKWMEQFEMAISNIYPENATANGHDFQMFSFEETTSCKACQMLLRGTFYQGYRCYRCRAPAHKECLGR VPPCGRHGQDFAGTMKKDKLHRRAQDKKRNELGLPKMEVFQEYYGIPPPPGAFGPFLRLNPGDIVELTKAEAEHNWWEGR NTATNEVGWFPCNRVHPYVHGPPQDLSVHLWYAGPMERAGAEGILTNRSDGTYLVRQRVKDTAEFAISIKYNVEVKHIKI MTSEGLYRITEKKAFRGLLELVEFYQQNSLKDCFKSLDTTLQFPYKEPERRAISKPPAGSTKYFGTAKARYDFCARDRSE LSLKEGDIIKILNKKGQQGWWRGEIYGRIGWFPSNYVEEDYSEYC
- Start the search ... and check the mails with a web browser. How many motifs are found? Which one of the motifs is common to all the 6 sequences?
- Can you see any link between the motifs found by MEME and domains in Pfam (paste a sequence in the Pfam Protein search service and look at the results). Can you describe the motifs found by MEME using the Pfam annotation?
Tricks: The result will be sent by e-mail, and it could take a bit long, so just move to the next exercise until you get the result.
10.- Genomics
Practice 10.1: Mitochondria of various organisms use different genetic code. Search the Internet site to obtain information for the mitochondrial codons from either vertebrates or invertebrates. Discuss usage differences between the standard codons and mitochondrial codons.
Practice 10.2: Retrieve one each of the cytosolic tRNA specific for the following amino acids from Saccharomyces cerevisiae:(a) Aspartic acid, (b) Phenylalanine, and identify their anticodons.
Practice 10.3: Retrieve nucleotide sequences (in fasta format) and restriction maps for one each of bacterial plasmid, cosmid and shuttle vector.
Practice 10.4: Retrieve DNA sequence encoding for human glucagon mRNA. Subject the sequence to Webcutter and construct the restriction map using REBASE restriction enzymes.
Practice 10.5: Search the candidate primers of PCR for the human pro-optiomelanocortin gene with the following sequence:
_gi _4505948 _ ref _ NM—000939.1 _ Homo sapiens proopiomelanocortin (POMC) AGCGGCGGCGAAGGAGGGGAAGAAGAGCCGCGACCGAGAGAGGCCGCCGAGCGTCCCCGCCCTCAG AGAGCAGCCTCCCGAGACAGAGCCTCAGCCTGCCTGGAAGATGCCGAGATCGTGCTGCAGCCGCTC GGGGGCCCTGTTGCTGGCCTTGCTGCTTCAGGCCTCCATGGAAGTGCGTGGCTGGTGCCTGGAGAG CAGCCAGTGTCAGGACCTCACCACGGAAAGCAACCTGCTGGAGTGCATCCGGGCCTGCAAGCCCGA CCTCTCGGCCGAGACTCCCATGTTCCCGGGAAATGGCGACGAGCAGCCTCTGACCGAGAACCCCCG GAAGTACGTCATGGGCCACTTCCGCTGGGACCGATTCGGCCGCCGCAACAGCAGCAGCAGCGGCAG CAGCGGCGCAGGGCAGAAGCGCGAGGACGTCTCAGCGGGCGAAGACTGCGGCCCGCTGCCTGAGGG CGGCCCCGAGCCCCGCAGCGATGGTGCCAAGCCGGGCCCGCGCGAGGGCAAGCGCTCCTACTCCAT GGAGCACTTCCGCTGGGGCAAGCCGGTGGGCAAGAAGCGGCGCCCAGTGAAGGTGTACCCTAACGG CGCCGAGGACGAGTCGGCCGAGGCCTTCCCCCTGGAGTTCAAGAGGGAGCTGACTGGCCAGCGACT CCGGGAGGGAGATGGCCCCGACGGCCCTGCCGATGACGGCGCAGGGGCCCAGGCCGACCTGGAGCA CAGCCTGCTGGTGGCGGCCGAGAAGAAGGACGAGGGCCCCTACAGGATGGAGCACTTCCGCTGGGG CAGCCCGCCCAAGGACAAGCGCTACGGCGGTTTCATGACCTCCGAGAAGAGCCAGACGCCCCTGGT GACGCTGTTCAAAAACGCCATCATCAAGAACGCCTACAAGAAGGGCGAGTGAGGGCACAGCGGGCC CCAGGGCTACCCTCCCCCAGGAGGTCGACCCCAAAGCCCCTTGCTCTCCCCTGCCCTGCTGCCGCC TCCCAGCCTGGGGGGTCGTGGCAGATAATCAGCCTCTTAAAGCTGCCTGTAGTTAGGAAATAAAAC CTTTCAAATTTCACA
Suggest which of the candidate pairs lead to the synthesis of the constituent hormone(s) (adrenocorticotropin, _-lipotropin, _-melanocyte stimulating hormone, _-melanocyte stimulating hormone, and/or _-endorphin).
Practice 10.6: Use BioEdit to translate the human pro-optomelanocortin. Deduce the frame from which the constituent hormones are likely to be translated.
Practice 10.7: Retrieve complete CDS of human alcohol dehydrogenase (ADH) isozymes from UniGene and record the comparison with other organisms (UniGene listings).
Practice 10.8: The file F352SS4 has a sequence of a nucleic acid from a comercial automatic sequencer. Discarding the beggining and the end, get the DNA sequence. Check if there are any problem with nucleotide identification. If so, correct the error/s according to the peaks in the electropherogram. Use the program Chromas.
Practice 10.9: Producing a restriction map.
Use the DNA sequence of the gene that codes for protein P57727 and search for enzymes that cut a minimum of once and a maximum of twice, and have a recognition site length of at least six bases.
Tricks: Use the program WEBCUTTER.
Practice 10.10: Translation.
Retrive the complete sequence entry for the DNA sequence of the gene that codes for protein P57727. Which part of the DNA sequence correspond to the coding sequence (CDS)? Do you see a difference between the annotated CDS and the predicted one? How can you explain this?
Tricks: Use the program APE and translate the coding sequence to the corresponding protein product. Report only ORF with a minimum nucleotide length of 300 nucleotides.
Practice 10.11: Designing primers.
Design the 6 best primers for the DNA sequence of the gene that codes for protein P57727. How many primers pairs are considered OK by the program? Design again primers for the sequence, but since you suspect vector contaminations, exclude the first and the last 12 base pairs of the sequence. Design an internal oligo to detect one of the sequence variants listed in the protein sequence entry P57727.
Tricks: Use the program APE and OLIGOCALC or NEBCUTTER.
11.- Proteomics
Practice 11.1: A purified mammalian protein with a molecular weight of 40,000±1000 Da and a pI of 8.0±0.5 is composed (an amino acid composition in mol %) of Ala, 7.5; Arg, 3.2; Asn, 2.1; Asp, 4.5; Cys, 3.7; Gln, 2.1; Glu, 5.6; Gly, 10.2; His, 1.9; Ile, 6.4; Leu, 6.7; Lys, 8.0; Met, 2.4; Phe, 4.8; Pro, 5.3; Ser, 7.0; Thr, 6.4; Trp, 0.5; Tyr, 1.1; and Val, 10.4. Try to identify the protein. [solution]
Tricks: Use Expasy tools AACompIdent.
Practice 11.2: Analyze peptide fragments produced by
treating a protein with the following amino acid sequence with
chymotrypsin, proteinase K, and trypsin. Deduce the specificities of
these enzymes.
_gi_35497_emb_CAA78820.1_ protein kinase C gamma [Homo sapiens] QLEIRAPTADEIHVTVGEARNLIPMDPNGLSDPYVKLKLIPDPRNLTKQKTRTVKATLNPVWNETFVFNL KPGDVERRLSVEVWDWDRTSRNDFMGAMSFGVSELLKAPVDGWYKLLNQEEGEYYNVPVADADNCSLLQK FEACNYPLELYERVRMGPSSSPIPSPSPSPTDPKRCFFGASPGRLHISDFSFLMVLGKGSFGKVMLAERR GSDELYAIKILKKDVIVQDDDVDCTLVEKRVLALGGRGPGGRPHFLTQLHSTFQTPDRLYFVMEYVTGGD LMYHIQQLGKFKEPHAAFYAAEIAIGLFFLHNQGIIYRDLKLDNVMLDAEGHIKITDFGMCKENVFPGTT TRTFCGTPDYIAPEIIAYQPYGKSVDWWSFGVLLYEMLAGQPPFDGEDEEELFQAIMEQTVTYPKSLSRE AVAICKGFLTKHPGKRLGSGPDGEPTIRAHGFFRWIDWERLERLEIPPPFRPRPCGRSGENFDKFFTRAA PALTPPDRLVLASIDQADFQGFTYVNPDFVHPDARSPTSPVPVPVM
Tricks: Use the utility PeptideMass from ExPASy)
Practice 11.3: Given the following amino acid sequence, estimate its amino acid composition, numbers of charged residues,
extinction coefficient, estimated half-life, and instability index of
the protein. Elaborate briefly how extinction coefficient, half-life and
instability index are estimated.
_gi_68532_pir__SYBYDC partate--tRNA ligase (EC 6.1.1.12), cytosolic - yeast MSQDENIVKAVEESAEPAQVILGEDGKPLSKKALKKLQKEQEKQRKKEERALQLEAEREAREKKAAAEDT AKDNYGKLPLIQSRDSDRTGQKRVKFVDLDEAKDSDKEVLFRARVHNTRQQGATLAFLTLRQQASLIQGL VKANKEGTISKNMVKWAGSLNLESIVLVRGIVKKVDEPIKSATVQNLEIHITKIYTISETPEALPILLED ASRSEAEAEAAGLPVVNLDTRLDYRVIDLRTVTNQAIFRIQAGVCELFREYLATKKFTEVHTPKLLGAPS EGGSSVFEVTYFKGKAYLAQSPQFNKQQLIVADFERVYEIGPVFRAENSNTHRHMTEFTGLDMEMAFEEH YHEVLDTLSELFVFIFSELPKRFAHEIELVRKQYPVEEFKLPKDGKMVRLTYKEGIEMLRAAGKEIGDFE DLSTENEKFLGKLVRDKYDTDFYILDKFPLEIRPFYTMPDPANPKYSNSYDFFMRGEEILSGAQRIHDHA LLQERMKAHGLSPEDPGLKDYCDGFSYGCPPHAGGGIGLERVVMFYLDLKNIRRASLFPRDPKRLRP
Tricks: Use the utility ProtParam from ExPASy
Practice 11.4: Compare the hydrophobicity/polarity
profiles (%buried residues, % accessible residues) and presents the result in a profile plot for human
serine protease with the following amino acid sequence.
__gi_2318115_gb_AAB66483.1_ MKKLMVVLSLIAAAWAEEQNKLVHGGPCDKTSHPYQAALYTSGHLLCGGVLIHPLWVLTAAHCKKPNLQV FLGKHNLRQRESSQEQSSVVRAVIHPDYDAASHDQDIMLLRLARPAKLSELIQPLPLERDCSANTTSCHI LGWGKTADGDFPDTIQCAYIHLVSREECEHAYPGQITQNMLCAGDEKYGKDSCQGDSGGPLVCGDHLRGL VSWGNIPCGSKEKPGVYTNVCRYTNWIQKTIQAK
Tricks: Use the utility ProtScale from ExPASy, which computes amino acid scale (physicochemical properties/parameters)
Practice
11.5: Scan the relative mutability of the protein with the following amino
acid sequence:
__gi_67414_pir__ KDIPRCELVKILRRHGFEGFVGKTVANWVCLVKHESGYRTTAFNNNGPNSRDYGIFQINSKYWCNDGKTR GSKNACNINCSKLRDDNIADDIQCAKKIAREARGLTPWVAWKKYCQGKDLSSYVRGC
Is there any correlation between the relative mutability and polarity, average flexibility and/or average buried area of the amino acid residues?
Tricks: Use the utility ProtScale from ExPASy, which computes these properties.
Practice 11.6: Search the Web site to predict transmembrane topology of rhodopsin with the following sequence and compare the membrane-spanning regions with the hydrophobicity profiles
__gi_10720173_sp_ MNGTEGPFFYVPMVNTTGIVRSPYEYPQYYLVNPAAYAALGAYMFLLILVGFPINFLTLYVTIEHKKLRT PLNYILLNLAVADLFMVLGGFTTTMYTSMHGYFVLGRLGCNIEGFFATLGGEIALWSLVVLAIERWVVVC KPISNFRFGENHAIMGLAFTWTMAMACAAPPLVGWSRYIPEGMQCSCGIDYYTRAEGFNNESFVIYMFIC HFTIPLTVVFFCYGRLLCAVKEAAAAQQESETTQRAEKEVTRMVIMMVIAFLVCWLPYASVAWYIFTHQG SEFGPVFMTIPAFFAKSSSIYNPMIYICLNKQFRHCMITTLCCGKNPFEEEEGASTASKTEASSVSSSSV SPA
Tricks: Use the Web server DAS to discover transmembrane helices. Use the utility ProtScale from ExPASy for hydrophobicity.
Practice 11.7: Comparative studies showed that the human TRPV1 channel is a membrane protein containing 6 transmembrane (TM) helices. Now we want to predict the exact location of TM helices, as well as the loops that goes inside and outside the membrane. Confirm the results and give the TM locations.
>TRPV1_HUMAN gi|62901455 MKKWSSTDLGAAADPLQKDTCPDPLDGDPNSRPPPAKPQLSTAKSRTRLFGKGDSEEAFPVDCPHEEGEL DSCPTITVSPVITIQRPGDGPTGARLLSQDSVAASTEKTLRLYDRRSIFEAVAQNNCQDLESLLLFLQKS KKHLTDNEFKDPETGKTCLLKAMLNLHDGQNTTIPLLLEIARQTDSLKELVNASYTDSYYKGQTALHIAI ERRNMALVTLLVENGADVQAAAHGDFFKKTKGRPGFYFGELPLSLAACTNQLGIVKFLLQNSWQTADISA RDSVGNTVLHALVEVADNTADNTKFVTSMYNEILILGAKLHPTLKLEELTNKKGMTPLALAAGTGKIGVL AYILQREIQEPECRHLSRKFTEWAYGPVHSSLYDLSCIDTCEKNSVLEVIAYSSSETPNRHDMLLVEPLN RLLQDKWDRFVKRIFYFNFLVYCLYMIIFTMAAYYRPVDGLPPFKMEKTGDYFRVTGEILSVLGGVYFFF RGIQYFLQRRPSMKTLFVDSYSEMLFFLQSLFMLATVVLYFSHLKEYVASMVFSLALGWTNMLYYTRGFQ QMGIYAVMIEKMILRDLCRFMFVYIVFLFGFSTAVVTLIEDGKNDSLPSESTSHRWRGPACRPPDSSYNS LYSTCLELFKFTIGMGDLEFTENYDFKAVFIILLLAYVILTYILLLNMLIALMGETVNKIAQESKNIWKL QRAITILDTEKSFLKCMRKAFRSGKLLQVGYTPDGKDDYRWCFRVDEVNWTTWNTNVGIINEDPGNCEGV KRTLSFSLRSSRVSGRHWKNFALVPLLREASARDRQSAQPEEVYLRQFSGSLKPEDAEVFKSPAASGEK
Tricks: Use the Web server TMHMM to discover transmembrane helices.
Practice 11.8: Studies on tetramerization of human TRPV1 channel pointed to the C-terminal as the putative region responsible of the quaternary structure channel formation. Check the possibility of coiled-coil domains in TRPV1 and give exact (putative) locations. Make MSA with other members of the family (TRPV2,TRPV3, TRPV4, TRPV5 and TRPV6) and check the conservation of tetramerization domain.
Tricks: Use the server COILS at EmbNet. For the MSA, make a whole protein alignment; then focus in cytoplasmic C-terminal only (discard N-terminal and TM sequences) and realign.
Practice 11.9: Given the following sequence and using as many bioinformatic tools as you need, characterize repetitions, motifs or domains that the sequence may contain. Propose a hypothesis of activity.
>mistery MLQTGLAKFGSHFTEAEVQQLVNSTDVDKNGFIDYGEFNVFAQNLTIEEI KGIKNIFANLKDDNSGTITYPDLRSSMNQYGDPDHSVMSNIISEVDTDRD GKLKYDEFLTTSLEQNKMHQEDMLLKAFKHFDKDHNTFINLDESETGLIL DKSADDIRAAYTFARELGRGQLGVTYIVKDKSNGEFFACKSISQRKLRHT EEREDVRREIQIMRHLTSQPNLVNIKGGFEDKESVHLVMELCTAGEAFDR IIKRGHYSERAAADIFAGIVNVVDFCHLSGVMHRKLKPENFLFLNKHEDS LMKTTDFGVSVFIKQGRIYTEIVGSSYYVAPEVLQRSYGQELDMWSAGVI MYILLCGIPPFWAETERGIAEAILKGDLDYQGEPWPGISNSAKPLLRRML EDDPSKRLTAAQMLPHPWI
Tricks: Identify the domains and treat separately. Make a MSA with BLAST homologues to identify conserved residues, and use PROSITE to predict the activity of the different elements.
Practice 11.10: Amphipatic helices, helical wheels and Leucine Zipper. [solution]
What are amphipatic helices? How do you recognise them?
>mystery MVVVAAAPNPADGTPKVLLLSGQPASAAGAPAGQALPLMVPAQRGASPEAASGGLPQARK RQRLTHLSPEEKALRRKLKNRVAAQTARDRKKARMSELEQQVVDLEEENQKLLLENQLLR EKTHGLVVENQELRQRLGMDALVAEEEAEAKGNEVRPVAGSAESAALRLRAPLQQVQAQL SPLQNISPWILAVLTLQIQSLISCWAFWTTWTQSCSSNALPQSLPAWRSSQRSTQKDPVP YQPPFLCQWGRHQPSWKPLMN
There is a Leucine Zipper domain in this sequence (where?). Can you discover which protein it is? Does it have any solved structure? Obtain a helical wheel presentation of the Leucine Zipper domain you visualized. Think about the function of the Leucine Zipper in the context of amphipaticity and hydrophobicity of the residues.
Tricks: Detect the presence of Leucine zippers in 2ZIP Server. Visualize the helical wheel by means of this helical wheel applet and in helical projections.
Practice 11.11: Find Protein domains/motifs
How many different domains are present in the Swiss-Prot entry P57727?. Is the information about the protein domains also annotated in the Features FT lines of the SwissProt entry file?
Tricks: Use the EMBOSS application patmatmotifs to search your sequence against the PROSITE motif database. Please select the output option of the program to provide full documentation for the matching patterns.
12.- Structural Bioinformatics
Practice 12.1: Editing structures with SwissPDB viewer. Structural alignments vs sequence alignment [solution]
Search for the following high resolution structures of SH3 domains: 1abo, 1awj, 1bb9, 1cka, 1csk, 1fmk, 1fyn. Load them with SwissPDB viewer and make the following:
Locate the structures corresponding to the SH3 domains and delete other proteins present in the file.
Change the name of the peptide chains to A.
Save the new structures. Save the sequences in fasta format.
Load the isolated SH3 domains and make an structural alignment.
Make a MSA with CLUSTALW using the fasta sequences.
Look at the sequence alignment and the structural alignment and compare. Are both alignments similar?
Tricks: Take the structures from ADAN database, either the original pdb files (1abo.pdb) or the cleaned ones (1abo2.pdb), having the SH3 domain already isolated. See manuscript pag. 5.
Practice 12.2: Homology Modeling. [solution]
The human transmembrane protease (Swiss-Prot
P57727)
consists of 3 domains: the LDL receptor domain, the SRCR domain and the
serine protease domain. Information about the location and the function
of the different domains, can be retrieved by consulting the InterPro
entry for that protein.
We are going to model the LDL domain of the protein, whose amino acid
sequence is:
>sp|P57727|TMS3_HUMAN_LDL HFDCSGKYRCRSSFKCIELIARCDGVSDCKDGEDEYRCVRV
1) First of all we are going to look for a suitable template, e.g. using
SAM HMM search algorithm accessible from the Tools section (Template
identification) of the
SWISS-MODEL Workspace. You can use the SWISS-MODEL Workspace
anonmously or as a registered user. If you would like to use the server
anonymously please remember to bookmark the page of the results. How
many different hits do you detect? How do they differ? To build the
model we will use the structure corresponding to the PDB entry
2gtl chain N as template. Save chain N of the pdb entry 2gtl locally on your
computer.
2) In the second step we are going to build an alignment between the
target and the template sequence. Multiple sequence alignments between
target, template and related sequences perform better than a simple
target/template pair-wise sequence alignmnet. Therefore we are going to
search for related sequences, e.g. by running a
BLAST
against the Swiss-Prot protein sequence database. Multiple sequence
alignment (MSA) of the LDL domain sequences can be then calculated using
the
T-Coffee MSA tool. Please be carful to align only the regions of the
different homologous sequences which correspond to the LDL domain.We
will use the information from this MSA to adjust the alignment between
our target sequence and the template (2gtlN).
3) Once we have obtained the MSA alignment we can prepare with the
help of DeepView a so called project file
containing the alignment between the target sequence and the template
sequence. This project file will be used by SWISS-MODEL to build a 3D
model for the LDL receptor of our target protein P57727.
Once you have obtained the model, please answer the following questions:
Are you sure that your model has the correct protein fold? Which
structural features are characteristic for this protein domain?
A protein sequence variant D(103)G has been described (D(103) of the
full length protein correspond to D(34) in the LDL domain). What do you
think is the function of the mutated residue in the native (not mutated)
protein domain? Do you think the mutation will affect the function of
this protein domain? Why?
Tricks: To produce a DeepView project file to be submitted
to the SWISS-MODEL homology modeling server please follow these steps:
- Load your target sequence (contained in a flat/text file):
DeepView-> SwissModel->Load Raw Sequence to Model...
- Load the template file (2gtl_N) into DeepView (File->
Open PDB File ...)
- Fit the target to the template ( Fit->Fit Raw Sequence)
- Carefully check the alignment (DeepView->Window->Alignment)
between your target and the template sequences. If needed, amend the
alignment (check: Prosite
Patterns location, disulfid bridges, use the information of a multiple
sequence alignment of related sequences, ... ).
- Then save a "Project" file (File->Save->Project...)
- And submit this file to the Project Mode of the SWISS-MODEL Workspace,
in the Modeling section of the server.
To answer the question abount functionality, superpose the model and the template structure (DeepView-> Fit-> Iterative magic Fit) and check the residues around the mutated residue.
Practica 12.3: Model Evaluation. [solution]
A 3D model of the Drosophila
UDP-glucose 4-epimerase protein has been generated by homology
modeling. The structure of the Human homolog protein
Q14376 has been used as template. The PDB ID for the template is
1ek5. Two different models,
Model1 and
Model2 have been obtained: they differ in the alignment between the
target and the template.
Evaluate the two models by checking the following criteria:
- Inside/Outside distribution of hydrophilic/hydrophobic residues;
- Ramachandran Plot of the model;
- Energy of the model (GROMOS force field: in DeepView: Tools->Compute Energy(Force Field));
- ANOLEA atomic mean force potential of the model;
Which of the model would you trust more and why?
Tricks: Use the force fields implemented in ANOLEA and in FoldX (local program)
Practice 12.4: Modeling proteins. We are interested in suggest mutations that break the activity of two PDZ domains in D. melanogaster: P31007 and O97111. For this we want to have a look at the structures. Are there already structures for this PDZ domains? If not, can you model them?
Tricks: See manuscript pag. 11. See SwissPDB viewer manual.
Practice 12.5: Pymol usage: Make mutations in a structure. See the structural states in a NMR structure.
Look at the PDZ domain 1be9.pdb structure by using Pymol, and make mutations that disrupt the binding to the peptide. Try mutate Thr7 to Trp
Load the NMR structure 1d5g.pdb and see the animation between the different states in the molecule
Tricks: See short manual of pymol. Colour by chain. Hide waters and hydrogens if present and activate mutagenesis in the Wizard menu. For the NMR animation set Movie/Frame rate to 5FPS.
Practice 12.6: Electron density and coordinate accuracy
In this exercise, you are going to analyze the structure of a fatty acid binding protein and the corresponding electron-density map [1fabp] obtained by x-ray crystallography. Please save these files locally on your hard drive before starting the exercise.
- Display the protein structure and identify regions with high B-factors
- Open the electron density map and display the map and
familiarize yourself with the handling of electron density maps (EDM). (Contouring
values, etc.)
- Can you identify regions with well defined and badly defined
electron density?
- Do these correlate with B-factors?
Identifying residues in electron density maps. Here you are going to analyze the structure of a protein and the corresponding high-resolution electron-density map [1hel]. Save these files locally on your hard drive.
- Display the protein structure including side chains.
- Open the electron density map and display the map and familiarize yourself with the handling of EDM maps. (Contouring values, etc.)
- Which side chains could you easily identify by the shape of the electron density?
- The following residue positions in your PDB-file need to be
filled with the correct side chains:
Ala 38, 59, 64 and 66. Please identify the correct side chain and insert the best fitting rotamer. Please justify your choice.
Tricks: Use SwssPDB viewer.
Practice 12.7: Recognize a fold.
[solution]
We are really interested in the following PDZ domains: protein CG6416-PE (isoform E), and the segment polarity protein dishevelled from D. melanogaster. We would like to use a threading server to check the possibility to have a reliable model.
Tricks: Isolate the PDZ and send both sequences to the threading server 3D-PSSM to recognize a fold from a sequence. Check carefully the sequence alignments and try to find structures with few insertions and deletions, high identity percentage and low E-values (if possible).
Practice 12.8: Secondary Structure Prediction.
[solution]
The amino acid sequence
of the E.Coli SufD protein (P77689),
a stabilizer of iron transporter is:
>sp|P77689|SUFD_ECOLI Protein sufD OS=Escherichia coli (strain K12) GN=sufD PE=1 SV=1 MAGLPNSSNALQQWHHLFEAEGTKRSPQAQQHLQQLLRTGLPTRKHENWKYTPLEGLINS QFVSIAGEISPQQRDALALTLDSVRLVFVDGRYVPALSDATEGSGYEVSINDDRQGLPDA IQAEVFLHLTESLAQSVTHIAVKRGQRPAKPLLLMHITQGVAGEEVNTAHYRHHLDLAEG AEATVIEHFVSLNDARHFTGARFTINVAANAHLQHIKLAFENPLSHHFAHNDLLLAEDAT AFSHSFLLGGAVLRHNTSTQLNGENSTLRINSLAMPVKNEVCDTRTWLEHNKGFCNSRQL HKTIVSDKGRAVFNGLINVAQHAIKTDGQMTNNNLLMGKLAEVDTKPQLEIYADDVKCSH GATVGRIDDEQIFYLRSRGINQQDAQQMIIYAFAAELTEALRDEGLKQQVLARIGQRLPG GAR
The crystal structure of the protein has been solved (1vh4.pdb) and the results of secondary structure prediction programs can be compared with the actual 3D data. Compare the results you obtain from a first generation (e.g. GOR I) and a third generation (e.g. PHD) program of secondary structure prediction with the structure based secondary structure assignment (e.g. using STRIDE). What can you say about the number of correct predicted secondary structure elements? About their lenght?
Tricks: The protein is a dimer of two identical chains. Download the file from the PDB database web site and save chain A in a separate file. PHD is form the PredictProtein server (You have to first register to the server with your e-mail address. warnings: use the beta submission (faster!!) and in the prediction type option select only PHDsec). Other servers are PSIPRED, JPRED and PROF
Practice 12.9: Predict secondary structure elements. [solution]
Predict now
the secondary structure elements (e.g. with PHD) of the Yonk Protein (O31947)
and compare the results with the actual 3D data (2h4o.pdb).
Compare the accuracy of the prediction of secondary structure elements
of the Yonk protein with the one of the SufD protein (previous practice
5). Which one is more
accurate? Can you explain why?
Tricks: As before save chain A from 2h4o.pdb in a separate file. Hint: Compare the results of a Blast search of the SufD and of the Yonk proteins. How many homologous family members are detected by Blast for the two proteins? To run Blast you can go to the corresponding Swiss-Prot entry of the proteins: P77689 and O31947, and click on the button "Quick BlastP search" on the upper right corner of the website.
Practice 12.10: Disorder Prediction. [solution]
The sequence of the hypothetical protein Q9JY98 from Neisseria meningitidis (a bacteria) is:
>tr|Q9JY98|Q9JY98_NEIMB Putative uncharacterized protein OS=Neisseria meningitidis serogroup B GN=NMB1681 PE=4 SV=1 MTQETALGAALKSAVQTMSKKKQTEMIADHIYGKYDVFKRFKPLALGIDQDLIAALPQYD AALIARVLANHCRRPRYLKALARGGKRFDLNNRFKGEVTPEEQAIAQNHPFVQQALQQQS AQAAAETLSVEAEAAESSAAE
Its structure has been recently solved by X-ray. If you analyze the PDB file you realize that some atom coordinates corresponding to the residues: 1-21 and 114-141 are missing, indicating a region of native disorder in the protein. Compare it with the region of disorder found in the structure. How accurate is the prediction for this protein?
Predict now the disorder regions of the hypothetical protein yvyC from Bacillus subtilis P39737.
>sp|P39737|YVYC_BACSU Uncharacterized protein yvyC OS=Bacillus subtilis GN=yvyC PE=1 SV=1 MNIERLTTLQPVWDRYDTQIHNQKDNDNEVPVHQVSYTNLAEMVGEMNKLLEPSQVHLKF ELHDKLNEYYVKVIEDSTNEVIREIPPKRWLDFYAAMTEFLGLFVDEKK
The analysis of its structure reveals that there is a region of disorder at residues 23-35. How accurate is the disorder prediction for this protein? Is it better or worse than for the previous protein? Can you explain why?
Tricks: Compute the disorder prediction with the programs DISOPRED2 and GLOBPLOT.
13.- Creación de una base de datos y presentación en formato web
- Software necesario para llevar a cabo esta práctica.
- python actualizacion.
- PDB ejemplo: 1ad5.pdb - 1ad52.pdb (SH3), 1ad53.pdb (SH2), 1ad54.pdb (kinasa), 1YIU2.PDB (WW), 1TE02.PDB (PDZ).
- El objetivo es crear una base de datos en formato MS Access que contenga información sobre distintos dominios modulares proteicos implicados en interacciones proteína-proteína. Los dominios elegidos son WW, SH3, SH2, PDZ. Cada registro de esta base debe representar una estructura (X-ray o NMR) que la base de datos SMART haya clasificado como perteneciente a alguno de estos 4 dominios. Las distintas estructuras que SMART nos indica como pertenecientes a un tipo o familia de dominio modular pueden contener en un mismo fichero PDB más de un dominio, luego debemos identificar y aislar la estructura de nuestro interés.
- Por ejemplo un cristal de una kinasa puede contener el dominio catalítico de la enzima, además de un dominio SH3 y otro SH2. De este modo, nuestra base de datos debe presentar como registros aislados la estructura de ambos dominios.
- Para un registro dado, nuestra base debe recoger la siguiente información almacenada en distintos campos:
- Número de registro.
- Fecha de inclusión de este registro en la base de datos.
- Nombre de la entrada en el Swiss-Prot de la proteína completa que contiene ese dominio: ej. SRC_RSVSR y su link correspondiente a esta base, podemos usar la aplicación portable FrontPage.
- Primary accession number de Swiss-Prot de la proteína completa que contiene ese dominio: ej. P00524 y su link correspondiente.
- Secuencia en código FASTA de la proteína completa que contiene ese dominio: FASTA format
- Link a la base MINT asociado al nombre de acceso en Swiss-Prot para la proteína completa que alberga ese dominio: Q9VCU7
- Nombre del fichero PDB original que contiene la estructura/s del dominio/s de interés, con un link a ese fichero PDB.
- Nombre del fichero PDB extraído del PDB original y que únicamente contiene una estructura de un dominio dado. Como nomenclatura podemos usar el nombre del PDB original al que añadamos un numero de 2 en adelante: 1sps.pdb vs 1sps2.pdb
- Nombre de la Familia del dominio y link externo a una descripción del mismo en Pfam: SH3
- Nombre de la Familia del dominio y link externo a una descripción del mismo en S.M.A.R.T.: SH3
- Técnica experimental con la se ha obtenido la información que aporta el fichero PDB de cada registro de nuestra base de datos: X-RAY DIFFRACTION.
- De qué especie procede la proteína cuyo dominio hemos incluido en nuestra base de datos: ej. Rous sarcoma virus.
- Resolución atómica de la estructura resuelta en Å: ej. 2.7
- Título asignado por los autores que depositaron un fichero PDB dado de cada uno de los dominios que nos interesa en el Protein Data Bank: ej. BINDING OF A HIGH AFFINITY PHOSPHOTYROSYL PEPTIDE TO THE SRC SH2 DOMAIN: CRYSTAL STRUCTURES OF THE COMPLEXED AND PEPTIDE-FREE FORMS
- Referencia numérica del artículo publicado en alguna revista científica (si existe) para describir el fichero enviado al Protein Data Bank: 7680960 con un link a ese paper en MedLine.
{kind=link}
- Cuando generamos una base de datos y trabajamos en una institución académica sin ánimo de lucho, nuestra intención última es que esta sea accesible a la comunidad científica internacional. En la actualidad, el modo más fácil es albergar dicha base de datos en una página WEB y por lo que debemos construir los motores en código HTML, PHP, ASP, etc para que los usuarios naveguen con facilidad por nuestra base y encuentren la información que buscan.
- A este respecto, han de generarse scripts en código ASP que albergados en un servidor WEB que ejecute este código, junto con la base de datos que hemos generado con Microsoft Access, nos permita explorar la base que hemos construido. El IBMC tiene distintos servidores web que usaremos a lo largo de esta práctica para ejecutar el código creado. Nos vamos a ayudar del programa ASPmaker para generar los scripts en código ASP. Los alumnos han de instalar este programa y aprender a usarlo: es muy intuitivo.
- Debemos identificar nuestra base de datos con un NOMBRE. A ser posible corto y sonoro, fácil de recordar y que haga alusión al contenido de la información.
- Se recomienda que esta práctica sea realizada en dos grupos, en función de los alumnos matriculados en este curso.
- A modo de ejemplo, en la siguiente figura se muestra una posible visualización de la página web que hemos de construir (generando código ASP con el programa ASPmaker) para ver y obtener información de nuestra base de datos:
-
- En cada línea de esta tabla dinámica se muestra información de estructuras que son dominios SH3 y a la izquierda de cada registro hay una lupa que "amplia" la información asociada a un registro dado. En esta figura podremos observar que de todos los campos que hemos recogido en nuestra base de datos para un registro dado, hemos seleccionado 8 en frontal de la visualización de la base en la página web. El link que amplia la información debe presentar los datos de todos los campos para ese registro.