PROTEIN ENGINEERING
Outline
Molecular edition with DeepView and Yasara. Structural alignment. Homology modeling of SH3 domains. Model evaluation.
Installing software
The specific software used along these practices is linked in the corresponding exercises. Some of them are portable application, but others may require typical installation. The software is optimized to run under Windows XP with Java Runtime Machine plug-in installed.
Exercises
1.- Structure edition and alignment
Practice 1.1: Editing structures with DeepView/SwissPDB viewer 4.0
Install the DeepView program in your computer (Windows and Mac). Just unzip file on a folder called spdbv4/. No extra installation is needed.
Launch the program and download the SH3 structure '1abo.pdb' from internet (File/Import). Visualize the structure. Rotate by pressing left-mouse button; translate with right-mouse button. Center with upper-left icon.
Locate the structures corresponding to the SH3 domains and delete other proteins/chains present in the file. For '1abo.pdb' make the following:
- Color/Chain will color the different chains. There are two identical SH3 domains in complex with ligand.
- Open Control Panel (Wind/Control Panel). In Control Panel select chain B (left-click on any B letter) and Build/Remove selected residues. Idem with chain D. In Control Panel select the sulfate molecule SO4410 (at the end of the list) and delete it.
- Chain C is the ligand. Change the name of the peptide chain from C to B. Select chain C and press Edit/Rename current layer. Under Rename Chain... write B and press OK.
- Save the new structure (File/Save/Current layer). Name it as '1abo2.pdb'
Save 'only' the sequence SH3 in fasta format:
- Remove the ligand by selecting chain B and delete it.
- Save the sequences in fasta format (File/Save/Sequence (FASTA)...) and give an appropriate name (i.e.: '1abo2_seq.txt')
Close DeepView. Check your output results
Tips & Tricks: Take the structures from ADAN database, either the original pdb files (1abo.pdb) or the cleaned ones (1abo2.pdb), having the SH3 domain already isolated. See manuscript pag. 5.
Practice 1.2: Make an structural alignment and compare with sequence alignment.
Search for the following high resolution structures of SH3 domains: 1awj, 1bb9, 1cka, 1csk, 1fmk, 1fyn. Take the simplified structures (field PDB2) from ADAN database.
Open a fresh session of DeepView. Load '1abo2.pdb', prepared previously. Load all downloaded structures in DeepView. Use File/Load PDB File.
Very important: remove ligands (if present) in all structures. Press Windows/Layers info (a new window opens, containing the layer information). Select the first layer in Layers Info window and remove the ligand from '1abo.pdb' by selecting chain B (in Control Panel) and removing residues. Idem for the second layer, third layer, etc.
Make an structural alignment:
- Select the second layer on Layers Info window.
- Fit/Magic Fit. Use: Backbone atoms only. Use 1abo as reference.
Repeat this step for third layer, fourth layer, etc. Use 1abo (the first layer) as reference in all cases.
Now we have an structural alignment. Visualize the structure-derived sequence alignment with Windows/Alignments (a new window appears) and pressing CTRL+G.
Save the alignment with File/Save/Alignment... as a text file.
Now make a Multiple Sequence Alignment with CLUSTALW2 at EBI, using the fasta sequences of all these SH3.
Look at the sequence alignment and the structural alignment and compare. Are both alignments similar?
Tips & Tricks: Take the fasta sequence of all these SH3 domains from ADAN database. Search in ADAN the SH3 domains (i.e.: 1abo) and press the magnifying glass (to the left). Download the txt file under Modular Domain Information/Sequence Domain.
2.- Homology Modeling
Practice 2.1: Modeling 6 different SH3 domains from yeast.
The two-hybrid experiments done with our favorite protein give several putative interacting proteins. All these proteins have at least one SH3 domain. It would be quite interesting to model the SH3 domains and propose a mechanism of interaction.
To model a protein, first of all we need to look for a template. This can be done using different strategies, tools and web servers. Here we propose the use of WU-BLAST2 at EBI, the SWISS-MODEL Workspace, and FoldX.
There are a lot of SH3 crystals in the protein structure databases. So, it is expected to find a suitable template for the 6 SH3 domains. In addition we have detailed information on SH3 architecture, motifs, sequence determinants, core positions, loop length, binding pocket properties, etc. These information can be used as rules to select the appropriate template:
* The three conserved residues in the pocket (WPY, marked with a $ in red) should match in the alignment, as well as the hydrophobic core
residues (marked with an asterisk, in blue).* No insertion or deletions in the loops forming the binding pocket (RT and n-Src). The distal loop can be modeled if necessary
* Conserved Gly positions (marked with a % in green)
* Aromatic and polar motif conservation in the RT-loop (marked with an & in black).
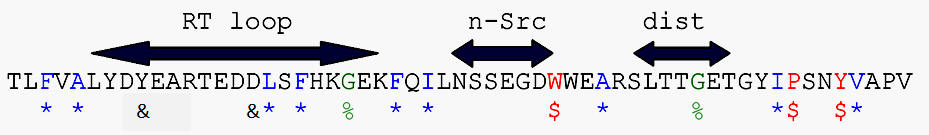
Example of alignment:
1fyn2 85
TLFVALYDYEARTEDDLSFHKGEKFQILNSS-EGDWWEARSLTTGETGYIPSNYVAPVD-
1abo2
85 NLFVALYDFVASGDNTLSITKGEKLRVLGYNHNGEWCEAQT-KNG-QGWVPSNYITPVNS
The SH3 sequences to be modeled are:
- >Sh3-1
YKAKALYPYDADDDDAYEISFEQNEILQVSDIEGRWWKARRANGETGIIPSNYVQLI- >Sh3-2
EYVEALYDFEAQQDGDLSLKTGDKIQVLEKISPDWYRGKSNNKIGIFPANYVKPA- >Sh3-3
RRVRALYDLTTNEPDELSFRKGDVITVLEQVYRDWWKGALRGNMGIFPLNYVTPI- >Sh3-4
GIYRAVYAYEPQTPEELAIQEDDLLYLLQKSDIDDWWTVKKRVIGSDSEEPVGLVPSTYIEEA- >Sh3-5
ETVTALYDYQAQAAGDLSFPAGAVIEIVQRTPDVNEWWTGRYNGQQGVFPGNYVQLN- >Sh3-6
GIVVAAYDFNYPIKKDSSSQLLSVQQGETIYILNKNSSGWWDGLVIDDSNGKVNRGWFPQNFGRPL
To proceed with the homology modeling, copy a query sequence and paste it in WU-BLAST2 at EBI.
- Start with Sh3-1
- In DATABASE select Protein and Protein Structure Sequence. This will search for homologue proteins which structures are already known. Other options remain as default.
- Run BLAST
- Check the E value (expectation value, see glossary) at the top of the list. The lower the E value, the more significant the score.
- Check the percentage of identity and similarity (glossary)
- Check the alignments of the top matches and have a look at the rules mentioned above for SH3 domains. Keep the selected alignment for later use
- DECIDE which template will be used for modeling. X-Ray better than NMR. Between X-Ray templates, choose the one with better resolution.
- Download the structure file from PDB RCSB or ADAN databases. Annotate whether the structure was determined by NMR or X-Ray (and the resolution in A)
- Perform the homology modeling by either FoldX or Swiss-Model Workspace as suggested below
Tips & Tricks:
For all SH3 modeling: Check the E values after BLAST, then check the alignments for percentages of identity and similarity, presence of gaps, motifs coincidence, etc.). Use WU-BLAST2 Protein at EBI or at NCBI
Sh3-1: Do you really need to model this SH3 domain?
Sh3-2: Choose the template with better resolution. Model with FoldX (see below).
Sh3-3: What about aromatic motifs? Model with Foldx (see below).
Sh3-4: What about gaps? Model with DeepView and send the job to SwissModel Workspace (see below).
Sh3-5: Check the results of the BLAST. Now we want a model refinement. To avoid the aromatic motif mismatch we will use a chimera made from two complementary templates: 1FYN.PDB and 1SSH.PDB. Download the project "fynssh.pdb" and build the chimera.
- Open the file in DeepView. You will see a superposition of 1FYN2.PDB and 1SSH2.PDB
- Select in layer 1 (1FYN2) the residues GLY30 to GLY55 (use Layers Info and Control Panel)
- Select in layer 2 (1SSH) the residues ASP56 to VAL90
- Click Edit/Create Merged Layer from Selection (by Layer). A new layer appears in Layers Info
- Select layer 3 (_merge_) and File/Save/Current Layer as i.e. "fyn-ssh.pdb"
- Save the sequence of the chimera and make an alignment with Sh3-5
Can you now use this file as template for modeling Sh3-5? If so, go ahead and model the domain with FoldX
Sh3-6: Try to find a suitable template... and good luck!
== Using FoldX for homology modeling ==
Prepare a folder for calculations. Save and unzip in this folder the file FoldX, containing teh executable "FoldX_28_26may08.exe", as well as the files "rotabase.txt", "template.txt", "mutant_file.txt", and the FoldX script "hmodel.txt"
Edit "template.txt" and write the name of the template pdb file (i.e. 1ABO2.PDB) in a single line. Save
Edit "mutant_file.txt" and write in the first line the template sequence, and in the second line the query sequence. Both sequences must be equal in lenght. No insertions or deletions are allowed. No symbols (i.e.: '-') different from single amino acid code letters are allowed.
Open a DOS shell window. Go to the folder where the executable FoldX lives and write:
>FoldX_28_26may08.exe -runfile hmodel.txt (and press enter)
['>' is the prompt of the operative system, so you don't need to type it]
After homology modeling calculations you will have finally your model (i.e. 1ABO2.PDB_1.PDB). Visualize the model with DeepView by superimposing the model with the template used for modeling. Check the alignment window.
After homology modeling calculations you will have finally your model (i.e. 1ABO2.PDB_1.PDB). Visualize the model with DeepView by superimposing the model with the template used for modeling. Check the alignment window.
== Using DeepView and the SWISS-MODEL Workspace ==
The modelling of a protein from its sequence against the known 3D structure of homologous protein(s) can be done with the help of DeepView by preparing a so called project file. This file contains the alignment between the target sequence and the template sequence. The project file will be used by SWISS-MODEL to build a 3D model for the target SH3 domains.
* Prepare your previously obtained alignment between the SH3 target sequence and the homologue pdb structure.
* Save your target sequence in FASTA format in a text file (i.e. “target.txt”).
* Choose the Load Load Raw Sequence from Amino Acids... tool from the SwissModel menu and open “target.txt”. The target sequence appears on the Display window as a long helix.
* Open the template structure of the reference molecule (File/Open PDB File...) i.e.: template.pdb.
* Click the little arrow beside the question mark of the Alignment window to view a plot of threading energies. Click smooth to set smooth to 1 and check SwissModel/Update threading display now tool. Select Color/By Threading Energy to display threading energy profile of the structure (the mean force potential energy of the polypeptide chain increases with colour varying from blue to red).
* Click SwissModel/Update threading display now tool. Select Color/By Threading Energy to display threading energy profile of the structure (the mean force potential energy of the polypeptide chain increases with colour varying from blue to red). Now the query protein is mounted on the template. Centre the molecule in the Display window.
* The alignment of the target sequence onto the template can be manually refined on the Alignment window by translating residues or inserting and removing gaps.
* Select a residue in the Alignment window and use CTRL+space bar to insert a gap or the CTRL+backspace key to remove a gap. When inserting a gap, a long bond appears in the Display window.
* Perform two operations: Select/Select_aa Making Clashes, and Tools/Fix Selected Side Chains/Quick and Dirty. Repeat the process to decrease the number of amino acid residues making clashes.
* Save the project File/Save/Project (All layers)... as i.e.: "proj_Sh3-1.pdb"
* Send the project for modeling to SwissModel Workspace. This web server can be used anonymously or as a registered user. We will try to use the server anonymously, so remember to bookmark the page of the results while processing. Alternatively you can register yourself and login in Workspace.
* In the Wokspace select Modeling/Project Mode. Fill in the e-mail and title of your work and upload your project file. Send the job to the server and wait for the results. Bookmark this page.
* Download the model as DeepView project and visualize it.
* Select the first layer and save the model as i.e. "Sh3-1_model.pdb". The file is ready for evaluation in terms of energy.
Practice 2.2: Model Evaluation.
The 3D models of SH3 domains have been generated by homology modeling by
using different templates.
Evaluate the models by checking the following criteria:
- Ramachandran Plot of the model. Are there residues with dihedrals in forbidden areas?
- Energy of the model with GROMOS force field.
- ANOLEA atomic mean force potential of the model.
- Stabilization energy calculated by FoldX
Which of the model would you trust more and why?
Tips & Tricks:
Ramachandran: Load the model in DeepView, select all residues (Select/All) and press Wind/Ramachandran.
GROMOS: In DeepView load the model and Tools/Compute Energy(Force Field). Take the energy value and compare with template energy
ANOLEA: Go back to your modeling results in SwissModel Workspace (the bookmarks) and select Anolea and/or Gromos and/or veryfy3d. For models obtained with FoldX, go to ANOLEA
FoldX: Measure the Gibbs energy of folding decomposed into different terms. Compare models with templates.
Use FoldX by preparing a folder with the executable "FoldX", the "rotabase.txt", "list.txt" and "Eanalysis.txt". Edit "list.txt" and type the name of model/s and template/s you want to analyze (one name per line).
In a DOS shell type: >FoldX_28_26may08.exe -runfile Eanalysis.txt (and press enter)
Check the output file "stability_energy.txt". Paste the content of the file in excel for convenience and try to answer the main question in the practice.
Practice 2.3: Energy minimization of structural models.
a) Take models Sh3-1.pdb to Sh3-4.pdb from practice 2.1 and minimize energy.
b) Re-evaluate energy and compare with energy obtained in practice 2.2.
Tips & Tricks:
Use GROMOS96 implemented in DeepView. Go to Prefs/Energy minimization and set the following parameters: 200 iterations steps of Steepest Descent and 200 iterations of Conjugate Gradients; Cutoff= 8.7. Be sure that Show Energy Report is activated. Load the model in Deepview. Go to Select/All to select all atoms and press Tools/Energy minimization (a progress bar appears) and read the energy value in the report/s. Save the minimized model with a different name, i.e.: "Sh3-1_min.pdb".
In addition download Pymol, decompress and install it. Open Pymol and load the model (File/Open...). In the menu windows press Builder. In the Builder menu press Fix button and select All C-Alphas. This will fix the alpha-carbons in order to avoid big changes in the protein folding. Now press Sculpt and wait for 30 s approximately. Then press Sculpt again to stop de proccess. Save the model with File/Save Molecule..., select the object describing your model and OK. Usa a different name, such as "Sh3-1_min-py.pdb".
Evaluate energy of all minimized models with methods stated in practice 2.2.
3. Yasara view. Just have a look
Practice 3.1: View yasara help movies.
Download and unzip Yasara_view (42MB) on a folder called i.e.: yasara_view
Run the executable"Yasara.exe"
Watch yasara movies: 1. General and 2. Molecular Graphics
Tips & Tricks:
Go to Help/Play help movie and select movies
Practice 3.2: Repeat practice 1.1 but using Yasara. Editing structures with Yasara
Open Yasara structure and
download the SH3 structure 1abo.pdb (go to “File/Load/Pdb File from
Internet” and enter“1abo” and press “Ok”). Visualize the structure:
- Switch view from CPK to wireframe (View/Scene Style/Stick)
- Rotate the structure by pressing the left-mouse button
- Translate the structure by pressing the middle-mouse button down (or with
both the left and right button pressed down).
- Zoom the right-mouse button pressed down
- Center with third-icon from the right
- Next we locate the structures corresponding to the SH3 domains, the SH3 ligands and other molecules (water and sulfate ions). Coloring by protein chains allows to easily visualize the different protein chains in 1abo. Go to “View/color/molecule” in the window on the right (“belongs to or has”), select “protein” and press “OK”, then select a color (e.g. blue) by clicking in the circle or in the list and press “Apply Color Gradient”, select a second color (e.g. red) and press “OK”. This colors the different protein chains. Note that there are two identical SH3 domains in complex with a ligand. Left click on the individual chains and check the left “on screen display” (OSD) panel for the chain ID (in the row labeled “residue” next to the amino acid identifier). (Press the “INS” or “Insert” button to toggle the OSD on/off) Molecules A and B correspond to the two SH3 domains and Molecules C and D belong to two (identical) SH3 ligands.
- For our modeling exercises we only need one SH3 domain and one ligand and we
will delete other proteins/chains/molecules present in the file. We will
keep chain A (SH3) and C (SH3 ligand) and remove the rest. Go to the right
OSD panel and left click “1abo”. A tree listing multiple “Molecule A-D’s”
entries unfolds: only the first 4 molecules contain the protein chains the
other copies contain water molecules and the sulfate ions. By toggling “yes”
to “no” in the “Vis” column you can easily check which molecule in the list
correspondents to the displayed molecule. To delete chain B, right click the
first “Molecule B” and select “Delete”. Repeat this for all “Molecules B and
D” and for the second 2nd and 3th entries of “Molecule A and C”. Check every
time that indeed the only the water and sulfate containing copies of
Molecule A and C are removed. In case the wrong “Molecule” entry is removed
go to “Edit/undo” or use the fourth icon from the left.
- Chain C is the SH3 ligand. Change the name of this peptide chain from C to
B. Right click chain C in the right OSD panel and select “Rename”, enter “B”
and press OK.
- Re-center the molecule by selecting “Edit/Center/All”.
- Save the new structure (File/Save As/PDBFile/ select 1abo), chose an
appropriate directory and name it 1abo2.pdb
- Save the sequence of the SH3 domain ( “File/SaveAs/Fasta Sequence/Molecule”),
select Molecule A, pick the same directory as before and give an appropriate
name (i.e.: '1abo2_A.fasta')
Tips & Tricks:
The ADAN database contains both the original pdb files (e.g. 1abo.pdb) or the cleaned ones (e.g. 1abo2.pdb), which have the SH3 domain (and—if present—the ligand) already isolated.